Advanced Therapeutic and Translational Science Department
At our Advanced Therapeutic and Translational Science Department, we provide a unique platform at the intersection of education, consultancy, and implementation. Our mission is to empower companies, startups, and academic institutions to accelerate the translation of innovative biomedical ideas into impactful therapies.
We integrate scientific excellence with practical expertise, offering tailored programs and solutions across the following domains:
AI-driven Drug Discovery and Design – Applying artificial intelligence to accelerate early-stage research and therapeutic innovation.
Drug & Gene Delivery – Developing knowledge and strategies for advanced delivery platforms.
Genetic Engineering – Enabling breakthroughs in genome editing, synthetic biology, and next-generation therapeutics.
Cell Processing and Functional Testing – Building competencies in cell therapy development, scale-up, and evaluation.
Quality & Regulatory Affairs – Guiding organizations to meet international compliance, safety, and efficacy standards.
Our approach is collaborative: we not only teach and consult but also co-create and implement solutions that drive progress in translational science and advanced therapeutics.
Vision:
To become a global partner of choice for biotech companies, startups, and academic institutions, enabling them to transform innovative ideas into real-world therapies that improve human health.
Mission:
- To deliver cutting-edge education and training in advanced therapeutic sciences.
- To provide strategic consultancy and practical solutions that support R&D, regulatory compliance, and translational pathways.
- To bridge academia and industry, fostering collaborations that accelerate innovation in drug discovery, genetic engineering, and cell-based therapies.
- To support startups and companies in building the capabilities needed to bring safe, effective, and high-quality therapeutics to patients.
AI-driven Drug Discovery and Design
Introduction to Drug Discovery and Design
Drug discovery and design lie at the heart of modern medicine, shaping the development of therapies that address unmet clinical needs across diverse diseases. The process begins with identifying a biological target—such as a protein, enzyme, or receptor—that plays a critical role in a disease pathway. Once the target is validated, researchers search for or design molecules capable of modulating its function, either to inhibit, activate, or fine-tune its activity. Traditionally, this relied heavily on serendipitous findings or labor-intensive screening of natural products and chemical libraries. Today, however, advances in molecular biology, high-throughput screening, and computational tools have made drug discovery a more systematic, rational, and predictive endeavor.
The design phase integrates medicinal chemistry, computational modeling, and structural biology to create drug candidates with optimal pharmacological properties. Rational drug design strategies leverage detailed knowledge of target structure and function to engineer molecules with higher specificity and potency. Complementing this, computer-aided drug design and AI-driven platforms accelerate the identification of novel chemical scaffolds, optimize drug-likeness, and predict ADMET properties before synthesis. Together, these approaches streamline the path from concept to candidate, reducing costs and timelines while improving the likelihood of clinical success.
Despite these advances, drug discovery and design face persistent challenges. One of the most significant is the extremely high attrition rate, as the majority of drug candidates fail during preclinical or clinical testing due to unforeseen toxicity or lack of efficacy. Another challenge lies in the complexity of human biology, where disease mechanisms often involve networks of interactions that are difficult to capture with single-target approaches. The rising costs and time demands of drug development also pose major barriers, as bringing a single drug to market can take more than a decade and billions of dollars. Data quality and reproducibility issues in both experimental and computational methods further complicate progress, while ethical considerations around personalized medicine, gene editing, and AI-driven drug design continue to emerge.
The workflow of drug discovery typically follows a stepwise path: target identification and validation, hit discovery through screening or design, lead optimization guided by structure–activity relationships, preclinical testing in cellular and animal models, and finally, clinical trials in humans. Each stage involves iterative refinement, where feedback from experiments and computational analyses is used to improve candidate molecules. Increasingly, interdisciplinary collaboration—between chemists, biologists, computational scientists, and clinicians—is recognized as essential for success in this complex process.
By combining traditional methods with cutting-edge computational and AI-driven innovations, modern drug discovery and design aim to make the process more efficient, cost-effective, and precise. While challenges remain, the integration of advanced tools and a deeper understanding of biology are steadily reshaping the field, bringing transformative therapies closer to patients worldwide.
Fundamentals of Artificial Intelligence and Machine Learning in Life Sciences
Artificial intelligence (AI) and machine learning (ML) have emerged as transformative forces in the life sciences, enabling researchers to analyze complex biological data, model intricate systems, and accelerate discovery in ways that were once unimaginable. At their core, these technologies rely on algorithms capable of detecting patterns, learning from large datasets, and making predictions that guide experimental design and therapeutic innovation. In life sciences, this often means integrating data from genomics, proteomics, metabolomics, medical imaging, and clinical records to uncover hidden relationships and actionable insights.
The fundamentals of AI and ML in this context include supervised learning, where algorithms are trained on labeled data to predict outcomes such as disease risk or drug response; unsupervised learning, which identifies natural clusters or patterns in biological datasets; and reinforcement learning, which iteratively improves decision-making strategies, such as in drug design or treatment optimization. Neural networks, particularly deep learning architectures, have demonstrated remarkable success in tasks like protein structure prediction, image-based diagnostics, and high-throughput compound screening. By automating analysis and learning from vast biomedical datasets, AI and ML empower researchers to move from raw information to biological understanding with unprecedented efficiency.
Applications are already reshaping research and healthcare. In diagnostics, AI models trained on imaging data detect tumors, cardiovascular anomalies, and neurological conditions with accuracy rivaling human experts. In genomics, ML tools identify disease-associated variants and predict the functional consequences of mutations. In drug discovery, AI accelerates target identification, molecular design, and ADMET prediction, shortening the timeline for therapeutic development. Even in personalized medicine, algorithms integrate genomic and clinical data to tailor treatments to individual patients, promising better outcomes and reduced side effects.
Yet, the integration of AI and ML into life sciences is not without challenges. One of the biggest obstacles is data quality—biological datasets are often noisy, incomplete, or biased, which can undermine model reliability. Interpretability remains another critical issue, as many AI models operate as “black boxes,” making it difficult for researchers and clinicians to understand the reasoning behind predictions. Computational cost is also significant, particularly for deep learning, which requires powerful hardware and substantial energy resources. Ethical concerns around data privacy, algorithmic bias, and equitable access to AI-driven healthcare further complicate adoption. Finally, validation and translation into clinical practice demand rigorous testing, as models must not only perform well in silico but also prove robust in real-world settings.
The workflow of applying AI and ML in life sciences often begins with collecting and curating large-scale datasets, ensuring that they are clean, representative, and well-annotated. Models are then selected or designed depending on the task, trained on subsets of the data, and validated against independent test sets to ensure generalizability. Successful models are further refined and integrated with experimental or clinical workflows, creating a feedback loop between computation and biology. With continuous advances in algorithms, data integration, and computational power, AI and ML are becoming indispensable tools for understanding life at every scale—from molecules to populations.
By mastering these fundamentals and addressing the challenges of data, interpretability, and ethics, the life sciences community is laying the foundation for a new era of discovery. AI and ML not only enhance research productivity but also hold the potential to reshape medicine, ushering in a future where biology and computation converge to deliver precise, efficient, and personalized healthcare solutions.
Data Sources and Drug Databases
In modern drug discovery and biomedical research, data sources and drug databases form the backbone of computational and experimental workflows. These resources provide structured collections of information about chemical compounds, biological targets, disease pathways, clinical outcomes, and pharmacological properties. By aggregating and standardizing vast amounts of data, drug databases allow researchers to identify patterns, make predictions, and guide decision-making at every stage of drug development. From early-stage target identification to late-phase clinical validation, access to reliable and comprehensive datasets ensures that researchers can build upon existing knowledge rather than starting from scratch.
Several widely used drug databases serve different but complementary purposes. For chemical and structural information, resources such as PubChem and ChEMBL offer millions of compounds with annotated biological activity data. DrugBank integrates chemical, pharmacological, and pharmaceutical data with clinical trial information, making it a cornerstone for translational research. Protein Data Bank (PDB) provides 3D structural information for proteins and complexes, essential for molecular docking and rational drug design. Other specialized databases like PharmGKB focus on pharmacogenomics, linking genetic variations to drug response, while ClinicalTrials.gov offers insights into ongoing and completed trials that inform clinical relevance. Together, these resources create a comprehensive ecosystem of knowledge that supports both academic and industrial research.
Despite their critical importance, several challenges limit the effective use of drug databases. One key issue is data quality—datasets may contain errors, inconsistencies, or missing values that reduce their reliability. Another problem is heterogeneity, as databases are often built using different standards, making integration across multiple resources difficult. Many databases also suffer from limited coverage, particularly for novel chemical space, rare diseases, or understudied targets. Data bias, especially toward well-studied molecules or diseases, can skew algorithmic predictions and hinder innovation. Accessibility and licensing restrictions further complicate matters, as some high-quality databases are proprietary and costly, limiting equitable access for academic researchers. Finally, keeping databases up to date with rapidly evolving biomedical knowledge remains a persistent challenge.
The workflow of using drug databases typically involves querying specific molecules, targets, or pathways, retrieving relevant datasets, and integrating them into computational pipelines such as virtual screening, molecular docking, or machine learning models. Researchers may also combine multiple data sources to cross-validate findings, improving robustness and reliability. Increasingly, AI and natural language processing tools are being applied to automatically mine biomedical literature and clinical data, expanding databases with new insights in real time.
By overcoming challenges of data quality, integration, and accessibility, drug databases and data sources will continue to serve as essential pillars of drug discovery. They not only accelerate innovation by providing researchers with curated knowledge but also enhance reproducibility and transparency in biomedical science, ensuring that future therapies are built on a foundation of reliable and comprehensive data.
AI-based Modeling of Molecular Structure and Function
Artificial intelligence has rapidly emerged as a transformative tool in computational biology, particularly in modeling the structure and function of biomolecules. By leveraging advanced algorithms, machine learning, and deep neural networks, AI enables the prediction of complex molecular conformations and interactions that were once difficult or impossible to determine with conventional experimental or computational methods. The breakthrough of AlphaFold in protein structure prediction has already reshaped structural biology, proving that AI can achieve near-atomic accuracy in modeling proteins. Beyond structural prediction, AI-based systems are being used to study ligand–receptor binding, enzyme kinetics, and conformational dynamics, offering a new level of insight into how molecules function at the atomic scale.
The applications of AI in molecular modeling extend across drug discovery, enzyme engineering, and personalized medicine. Pharmaceutical companies employ AI-driven platforms to accelerate virtual screening of drug candidates, reducing the time and cost required for preclinical development. In protein engineering, AI models assist in designing enzymes with enhanced stability or catalytic efficiency, while in genomics and precision medicine, AI helps predict the functional impact of mutations on protein folding and signaling pathways. These capabilities bridge the gap between raw sequence data and actionable therapeutic strategies, making AI indispensable for modern life sciences.
Despite its transformative potential, AI-driven molecular modeling faces several challenges. One major limitation is the dependence on large, high-quality datasets, as biased or incomplete data can result in inaccurate models. The “black box” nature of many AI algorithms also reduces interpretability, making it difficult to validate or explain predictions in a mechanistic way. Computational cost is another barrier, since training and deploying large-scale AI models require significant hardware resources. Furthermore, while AI has shown remarkable success in well-studied proteins and ligands, generalizing predictions to novel or underexplored biomolecular systems remains problematic. Finally, translating AI-generated insights into experimentally verifiable outcomes demands careful integration with laboratory workflows, which can slow adoption in industrial and clinical settings.
The process of AI-based molecular modeling typically involves several steps. Datasets containing structural or biochemical information are first collected and curated, often from protein databanks, chemical libraries, or high-throughput screening assays. These datasets are then used to train machine learning algorithms or deep learning architectures capable of recognizing patterns in molecular interactions. Once trained, the models can predict 3D structures, binding affinities, or dynamic behaviors of molecules with remarkable speed. Outputs are validated against experimental data or higher-level computational simulations such as molecular dynamics, ensuring reliability and accuracy. With continuous improvements in algorithm design, training strategies, and computational efficiency, AI-based molecular modeling is increasingly becoming a practical and scalable approach for guiding both basic research and therapeutic innovation.
By systematically addressing issues of data quality, algorithm transparency, and experimental integration, AI-based modeling holds the promise of revolutionizing structural biology, rational drug design, and personalized medicine. As the technology matures, it is poised to serve not only as a predictive engine but also as a creative partner in designing novel molecules with optimized structures and functions, ultimately reshaping the future of biomedical discovery.
Drug Design Using Generative Algorithms
Generative algorithms are redefining the landscape of drug discovery by enabling the creation of entirely new molecular entities rather than relying solely on existing chemical libraries. Unlike traditional approaches, which depend on screening millions of known compounds, generative models such as variational autoencoders (VAEs), generative adversarial networks (GANs), and reinforcement learning systems are capable of learning the “rules” of chemical space from large datasets and then producing novel molecular structures with desired properties. This ability allows researchers to explore vast, uncharted regions of chemical space, dramatically accelerating the search for drug candidates.
The applications of generative algorithms in drug design are already being realized across pharmaceutical research. These models can design molecules optimized for binding affinity, selectivity, solubility, or toxicity profiles, significantly reducing trial-and-error cycles. In oncology, for example, generative AI has been used to rapidly propose inhibitors for kinases and epigenetic regulators, while in infectious diseases it has generated candidate antivirals within weeks of target identification. Beyond small molecules, generative algorithms are increasingly applied to peptides and proteins, expanding their relevance to biologics and advanced therapeutics. By integrating predictive models for ADMET properties, these tools streamline the pipeline from hit identification to lead optimization.
Despite their promise, generative algorithms face important limitations. One major challenge is synthetic feasibility, as many AI-generated molecules may be chemically unstable or difficult to produce in the laboratory. Data bias presents another obstacle, since models trained on incomplete or non-representative datasets can fail to generalize, producing compounds that resemble existing chemistries rather than truly novel ones. Interpretability is also limited—understanding why an algorithm proposes a specific structure remains difficult, complicating decision-making for medicinal chemists. Computational cost can be substantial, particularly for reinforcement learning frameworks that require iterative optimization. Finally, the transition from virtual molecules to experimentally validated drug candidates remains resource-intensive, and discrepancies between in silico predictions and biological reality continue to pose risks.
The workflow of drug design using generative algorithms typically begins with large molecular datasets that are encoded into a latent chemical space. Generative models explore this space to propose novel compounds that meet predefined objectives, such as high predicted binding affinity or improved pharmacokinetics. These proposed molecules are filtered using predictive models and prioritized based on their drug-likeness and ADMET characteristics. Medicinal chemists then select top candidates for synthesis, which are experimentally validated in vitro and in vivo. Feedback from these experiments is fed back into the generative model, creating an iterative cycle that improves performance and relevance over time.
By systematically addressing challenges related to synthesis, interpretability, and validation, generative algorithms are poised to become central tools in drug discovery. Their capacity to rapidly generate, evaluate, and refine novel compounds opens the door to more efficient pipelines and innovative therapies, transforming the way new medicines are conceived and developed.
Target Identification with AI
Target identification is the first and perhaps most critical step in the drug discovery pipeline, as the success of an entire therapeutic program depends on selecting the right biological target. Artificial intelligence (AI) is revolutionizing this process by enabling researchers to analyze vast and heterogeneous datasets—ranging from genomics and transcriptomics to proteomics, imaging, and clinical records—with unprecedented depth and speed. AI models can uncover hidden correlations, identify disease-associated genes or proteins, and predict causal relationships that would be nearly impossible to detect with traditional bioinformatics. By integrating multiple omics layers, AI-driven approaches provide a systems-level understanding of disease mechanisms, guiding the selection of targets that are both biologically relevant and therapeutically actionable.
The value of AI-based target identification is already evident across therapeutic areas. In oncology, machine learning algorithms have been used to prioritize tumor-specific vulnerabilities by analyzing mutational signatures and expression profiles. In rare and complex diseases, AI can sift through noisy and limited datasets to highlight novel targets that were previously overlooked. Pharmaceutical companies are increasingly deploying these approaches to reduce the attrition rate in clinical development, ensuring that resources are invested in targets with a higher probability of success. Beyond drug discovery, AI-driven target identification also plays a role in personalized medicine, helping clinicians match therapies to patient-specific molecular profiles.
Despite its transformative potential, AI-based target identification faces important challenges. One major issue is data quality and availability—biomedical datasets are often incomplete, biased, or inconsistent, which can lead to misleading predictions. The interpretability of AI models is another concern, as black-box algorithms may suggest targets without providing mechanistic explanations, making experimental validation more difficult. Integration of heterogeneous data sources, such as combining omics with clinical outcomes, requires sophisticated computational pipelines and careful normalization. Reproducibility across laboratories and populations remains a critical hurdle, as models trained on specific cohorts may fail to generalize. Furthermore, the experimental validation of AI-predicted targets is resource-intensive and can limit the speed of translation to the clinic.
The workflow for AI-driven target identification typically involves curating large-scale datasets from public repositories, clinical trials, or proprietary studies. These datasets are preprocessed to remove noise and harmonize features before being fed into machine learning or deep learning models. Algorithms are then trained to detect associations between molecular features and disease phenotypes, producing ranked lists of candidate targets. These predictions are cross-validated with known biology and prioritized for downstream experimental studies, such as CRISPR screens or high-throughput assays. Iterative feedback between computational models and wet-lab experiments helps refine predictions, improving the likelihood of identifying robust, druggable targets.
By overcoming challenges related to data quality, interpretability, and validation, AI is establishing itself as a cornerstone in modern target discovery. Its ability to integrate diverse sources of biomedical information and highlight actionable insights not only accelerates the drug discovery process but also enhances precision medicine by ensuring that therapies are developed against the most promising molecular targets.
AI-enhanced 3D Structure Design and Molecular Docking
The integration of artificial intelligence into 3D structure design and molecular docking is transforming computational drug discovery. Traditionally, molecular docking has relied on approximations of binding interactions between ligands and targets, often limited by the accuracy of structural models and scoring functions. With AI-enhanced approaches, protein and ligand structures can be generated or refined at near-atomic resolution, and docking simulations are guided by deep learning models trained on large datasets of experimentally validated interactions. This synergy enables faster, more accurate prediction of binding affinities and poses, while simultaneously exploring larger chemical spaces than conventional docking tools could handle. AI not only improves docking precision but also accelerates the early stages of drug development by suggesting promising candidates with better confidence.
Applications of AI-driven docking are rapidly growing. Pharmaceutical companies employ these tools to virtually screen millions of compounds against difficult or novel targets, significantly reducing the cost and time of hit discovery. Beyond small molecules, AI-enhanced docking has been extended to peptides, antibodies, and nucleic acids, broadening its impact across therapeutic modalities. By coupling with AI-generated 3D protein structures—such as those predicted by AlphaFold—molecular docking can now address targets that previously lacked experimental structures. Moreover, reinforcement learning algorithms can iteratively optimize ligands to improve docking scores, creating a closed loop of design, evaluation, and refinement that accelerates lead optimization.
Despite these advances, several challenges remain. One major issue is the accuracy of scoring functions—AI models may improve predictions but can still misestimate binding free energies, particularly for flexible or allosteric sites. Data quality is another limitation, since training datasets often emphasize well-studied protein–ligand systems, leading to reduced performance on novel or less characterized targets. The interpretability of AI-driven docking results is limited, making it difficult for researchers to fully trust predictions without extensive validation. Computational cost is also a concern, as large-scale docking campaigns enhanced with AI still require significant GPU and CPU resources. Furthermore, translating docking predictions into experimental confirmation can be resource-intensive, and discrepancies between in silico and in vitro results remain a persistent challenge.
The workflow of AI-enhanced docking generally begins with high-quality structural data, either experimentally determined or AI-predicted. Molecules are encoded and fed into deep learning models that predict binding poses and affinities, often outperforming classical docking engines in speed and accuracy. Generative algorithms can propose new ligands, which are then iteratively docked and refined through AI-guided optimization. Top-ranked candidates are validated through molecular dynamics simulations and, eventually, experimental assays. This pipeline creates a powerful cycle of hypothesis generation and testing, greatly accelerating the transition from computational prediction to laboratory validation.
By addressing challenges related to scoring accuracy, dataset diversity, and experimental validation, AI-enhanced 3D structure design and molecular docking are poised to become essential pillars of modern drug discovery. These methods not only refine our understanding of molecular interactions but also open opportunities for designing innovative therapeutics with higher precision and efficiency.
Evaluation and Optimization of Drug Candidates
The evaluation and optimization of drug candidates represent critical phases in the drug discovery and development pipeline, bridging the gap between initial compound identification and preclinical or clinical validation. Once potential molecules are generated—whether through high-throughput screening, rational design, or AI-assisted methods—they must undergo rigorous assessment to determine their pharmacological potential. This involves a careful balance between efficacy, safety, and drug-like properties, as even the most promising molecules can fail if they lack suitable absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles. AI and computational models are increasingly being used to predict these properties early on, allowing researchers to prioritize compounds with higher chances of success.
Applications of drug candidate evaluation are wide-ranging. In vitro assays are used to test biological activity, selectivity, and cytotoxicity, while in vivo studies provide insights into pharmacokinetics and therapeutic potential. Computational methods complement these experiments by predicting solubility, permeability, and potential off-target effects, which accelerates decision-making and reduces the need for costly laboratory work. Optimization often involves modifying chemical structures to enhance potency, improve metabolic stability, or minimize toxicity. Iterative cycles of synthesis, testing, and computational refinement create a feedback loop that progressively transforms a preliminary hit into a viable lead compound.
Despite advances in methodology, significant challenges remain. One major issue is the high attrition rate, as the majority of drug candidates fail during preclinical or clinical phases due to unforeseen toxicity or insufficient efficacy. Another challenge lies in predicting complex human biology, since in vitro and animal models often fail to capture the full spectrum of human responses. Data quality and reproducibility are persistent concerns, as variability in assay conditions or computational models can lead to inconsistent results. Moreover, the optimization process itself can be resource- and time-intensive, requiring careful prioritization of which candidates to advance. Finally, translating computational predictions into experimentally confirmed outcomes remains a critical hurdle, as discrepancies between in silico models and real-world biology often emerge.
The workflow of evaluation and optimization typically begins with screening assays that assess biological activity and ADMET characteristics. Promising candidates are then subjected to structural modifications, guided by structure–activity relationship (SAR) studies and computational modeling, to improve their therapeutic index. Parallel computational simulations help predict the consequences of these modifications before experimental validation. This iterative process continues until a small number of optimized leads demonstrate both strong efficacy and acceptable safety profiles, making them suitable for advancement into preclinical development.
By systematically addressing challenges in prediction accuracy, reproducibility, and translational relevance, the evaluation and optimization of drug candidates ensure that only the most promising compounds progress through the costly stages of drug development. With the integration of AI and advanced computational tools, this process is becoming increasingly efficient, accelerating the delivery of safe and effective therapies to patients.
Drug & Gene delivery
Gene delivery vectors are engineered vehicles designed to transport therapeutic genetic material (DNA & RNA) into target cells to treat or prevent disease. They overcome the natural barriers cells have against foreign genetic material. Major vector classes include viral vectors (Biological) and Non-viral vectors (Synthetic/Chemical). The benefit of gene delivery vectors is that they are the essential engines driving gene therapy and genetic medicine. The choice between viral and non-viral systems involves a careful trade-off between efficiency, cargo capacity, duration, immunogenicity, safety, manufacturability, and cost.
Viral systems
Viral vectors exploit natural viral infection mechanisms. The advantages of these vectors consist of high efficiency of delivery, long-term expression, specific targeting, and well-studied and developed. The most common viral vectors are lentivirus, baculovirus, retrovirus, and adeno-associated virus (AAV). Most of these viral vehicles produce permanent expression, while adenoviruses create transient expression. Viral vectors are the go-to choice when long-term, high-level gene expression is required for a disease (e.g., hereditary disorders like hemophilia or SMA), and the risks of immunogenicity and mutagenesis can be managed.
Some vectors, like retrovirus, lead to the integration of the transgene into the host genome, which results in limitations in some activities, such as activation of oncogenes, insertional mutagenesis, packaging capacity of exogenous DNA, and neutralizing antibodies against AAV. To solve this problem, non-integrating vectors were designed to bypass activating oncogenes and achieve targeted integration by applying zinc finger nuclease (ZFN) to add the erythropoietin gene (Epo) into the chemokine (C-C motif) receptor-5 gene locus of cells.
Immunogenicity is another challenge in viral vectors that liver-targeted expression to deliver the gene to liver cells (hepatocytes) can promote tolerance to the transgene product. Using alternative technologies such as synthetic vectors like lipid nanoparticles (LNPs) or polymers to deliver the genetic material, mRNA COVID-19 vaccines are a famous example of LNP delivery. Gene editing, such as CRISPR-Cas9 system, instead of adding a new gene, aims to directly correct the mutation in the patient’s own DNA. Furthermore, since a large portion of the human population can neutralize AAV antibodies, the in vivo effect decreases dramatically. Hence, AAV vectors may be among the most promising vectors because of their reduced pathogenicity in humans and their ability to achieve long-term gene expression.
Packaging of viral vectors
Both AAV and lentivirus are produced using a method called transient transfection. Common procedures used for transfecting cells include chemical and physical methods. Chemical methods include polymeric carriers, lipid agents, inorganic nanoparticles, and dendrimers. Using inorganic nanoparticles (NPs) for transfecting cells is frequently associated with polycations. Lipid-Based Nanoparticles (LNPs) consist of cationic/ionizable lipids + helper lipids (phospholipids, cholesterol) + PEG-lipid encapsulating nucleic acids. Physical methods that directly perturb cell membranes include electroporation, nucleofection, gene gun, sonoporation, and hydrodynamic delivery. Choosing the transfection procedure or reagent is essential for increasing the transfection rate.
The fundamental principle is to transfer human embryonic kidney (HEK) 293 cells (or a derived cell line like 293T) with a set of plasmids that provide all the necessary components to make the virus, but are themselves unable to be packaged. The key feature of the packaging is its ability to infect both dividing and non-dividing cells by actively importing its genetic material into the nucleus. In the packaging procedure, replication-incompetent virus (safe for use) is used. For this reason, the viral genome is split across multiple plasmids to prevent the creation of a replication-competent virus.
There are three groups of plasmids in lentiviral packaging, which consist of the transfer plasmid (interest gene), packaging plasmids(Often split into two plasmids), and envelope plasmid.
The packaging procedure started by culturing HEK 293T cells (chosen for high transfection efficiency) to ~70-80% confluency. Transfection via co-transfection of the cells with the three (four) plasmids at an optimized ratio (e.g., Transfer, Packaging, and Envelope packaging 2:1:1) using different transfection reagents like PEI, calcium phosphate, and lipofectamine. Incubation of the cells to take up the plasmids and use their own machinery to express the viral proteins.
Virus assembly is done for the viral proteins and genomic RNA is assembled at the cell membrane and buds out. Harvesting step is done 48 and 72 hours post-transfection, the culture medium (containing the viral particles) is collected. Concentration & Purification is an important step where the medium is centrifuged at low speed to remove cell debris. The virus in the supernatant is then concentrated (often by ultracentrifugation) and/or purified (using chromatography or gradient centrifugation). Titration of the concentrated virus is quantified (titered) to determine its concentration (e.g., Transducing Units per mL – TU/mL) using methods like qPCR (genomic titer) or functional assays on target cells (functional titer). Storage of viral particles at -80°C.
One of the major challenges in the viral transfection procedure consists of delivery efficiency and cytotoxicity. There are some procedures for solving this problem, such as optimizing reagent-to-DNA ratios, using next-generation transfection reagents that are specifically formulated for viral production, and selecting alternative methods, including Calcium Phosphate Transfection: A classic, low-cost method that works very well for HEK 293 cells. It requires precise timing and pH control but can yield high titers with minimal cost. PEI (Polyethylenimine) Transfection: A very popular, cost-effective polymer for large-scale viral vector production. High-quality, linear PEI (e.g., PEIpro®) is an industry standard due to its good efficiency and scalability. Baculovirus/Sf9 System (for AAV): For certain vectors like AAV, using insect Sf9 cells infected with recombinant bacmams (baculoviruses carrying the necessary genes) can be more scalable and productive than mammalian cell transfection.
The choice between using lentiviruses or AAV depends on the experimental skill. For example, lentiviruses are used for integrating into difficult-to-transfect cells (like stem cells) or for long-term expression in dividing cells. AAV is used for efficient in vivo gene delivery to non-dividing cells with a superior safety profile.
Non- viral systems
Non-viral systems are preferred for applications where transient expression is sufficient (e.g., cancer therapy, vaccines, CRISPR gene editing), safety is the paramount concern, or a large gene needs to be delivered.
Advantages of non-viral vectors consist of low immunogenicity, no risk of insertional mutagenesis, large cargo capacity, ease of manufacturing and cost-effectiveness, and flexibility and design control.
Limitations of non-viral vectors include low transfection efficiency, which can be modified by incorporating cell-penetrating peptides (CPPs), targeting ligands, and nuclear targeting in the cargo to facilitate nuclear entry, transient gene expression that can be improved by utilizing CRISPR/Cas9 or transposon systems (e.g., Sleeping Beauty) for targeted genomic integration and engineered mRNA with modified nucleosides for enhanced stability and prolonged expression, cytotoxicity that can be overcomed by biodegradable polymers and natural & less toxic lipids, and difficulty with in vivo delivery that can be modified by polyethylene glycol (PEG) or other hydrophilic polymers, active targeting peptides, or antibodies, and physical methods such as electroporation, sonoporation, gene guns, and direct injection.
Non-viral transfection procedure
Transfection process is used to transfer non-viral vectors, which is achieved through chemical or physical methods. The transfection procedure has an exact protocol that varies by method and cell type, but the general workflow is consistent, which consists of cell seeding, transfection complex formation (Day of Transfection) that is the most crucial step to mix the nucleic acid with the transfection reagent to form stable, positively charged complexes called polyplexes (with polymers) or lipoplexes (with lipids), delivery to cells for adding complexes to cells, post-transfection & analysis through media changing, incubation and expression, and analysis for evaluation based on microscopy, flow cytometry, qPCR/RT-PCR, western blot or ELISA.
Some of the important challenges in the non-viral transfection procedure consist of low cellular uptake, which is reported because the negatively charged cell membrane repels the naked nucleic acid. Hence, modification of surface charge and creating vectors with a slightly positive charge (cationic) promotes interaction with the negative cell membrane. Conjugating specific molecules (e.g., antibodies, peptides, folate, carbohydrates) to the vector’s surface that bind to receptors on the target cell is another solution.
Endosomal entrapment and degradation is the biggest bottlenecks. Most of the vectors are taken up by the cell via endocytosis, becoming trapped in an endosome and degraded. So, polymers like PEI with a high buffering capacity absorb protons (H+ ions) in the acidic endosome and cause chloride ions (Cl-) to flood in. Moreover, using endosomolytic agents, such as incorporating peptides or other agents that are activated at low pH to disrupt the endosomal membrane.
Electroporation
Electroporation is a common procedure of transfection in non-viral vectors. Some of the challenges and solution are mentioned here. Low efficiency is overcome by systematically optimizing voltage, pulse length, MOI, and cell concentration. Damaging the virus can be overcome by using low-ionic-strength buffers and keeping samples cold. Low cell viability that can be overcome by optimizing pulse parameters (lower V, shorter time). Buffer incompatibility requires exchanging the buffer and using specialized electroporation buffers.
Our team has hands-on experience in addressing these pain points. We provide consulting services for selecting proper gene delivery vectors, improving the transfection procedure, training in advanced preparation techniques for gene targeting, and strategic guidance for increasing transfection rate, using the nuclease procedures, and regulatory compliance.
By tackling laboratory challenges, we help ensure gene delivery products reach their full potential in clinical and commercial use.
Liposomes
Liposomes are spherical vesicles composed of natural or synthetic phospholipids, widely recognized as one of the most reliable and clinically validated drug delivery systems. They have the unique ability to encapsulate both hydrophilic and lipophilic molecules, making them a versatile platform for pharmaceutical and biomedical applications. Their importance lies in protecting sensitive drugs, such as nucleic acids or peptides, from degradation while improving bioavailability and reducing systemic toxicity. Without proper formulation, however, liposomes may face issues such as poor encapsulation efficiency, drug leakage, or instability—directly affecting therapeutic success.
One of the greatest advantages of liposomes is their strong clinical track record; FDA-approved liposomal drugs like Doxil® demonstrate their potential for real-world impact. Surface modifications such as PEGylation or ligand attachment further enhance circulation time and allow targeted delivery. Liposomes have been successfully applied in cancer chemotherapy, antifungal treatments, vaccine development, and even cosmetic formulations.
Despite their promise, liposomes come with well-documented challenges. Shelf-life instability due to lipid oxidation and hydrolysis is common, but can be mitigated with antioxidants, controlled storage, or lyophilization using cryoprotectants. Drug leakage during storage and transport is another issue that often frustrates researchers; fine-tuning lipid composition and bilayer rigidity is essential to minimize this. In production, particle size heterogeneity often leads to unpredictable pharmacokinetics, requiring extrusion or microfluidic-based methods for tight size control. From an industrial perspective, high production costs and difficulties in scale-up are frequent barriers, particularly when moving from academic labs to GMP facilities. These can be addressed through continuous manufacturing systems, process automation, and careful raw material selection.
Several established methods are available for liposome preparation, each offering distinct advantages depending on the intended application, drug type, and scalability:
1. Thin Film Hydration (Bangham Method):
The classical and most widely used approach.
Lipids are dissolved in an organic solvent, dried as a thin film, and then hydrated with an aqueous buffer.
Produces multilamellar vesicles (MLVs) that can be downsized by sonication or extrusion.
2. Reverse-Phase Evaporation:
Lipids are dissolved in organic solvents and emulsified with an aqueous phase, followed by solvent removal.
Yields large unilamellar vesicles (LUVs) with relatively high encapsulation efficiency for hydrophilic drugs.
3. Ethanol or Ether Injection:
Lipid solution in a volatile solvent (ethanol or ether) is rapidly injected into an aqueous phase.
Results in small unilamellar vesicles (SUVs).
Simple and reproducible, but limited by solvent toxicity and removal challenges.
4. Microfluidics-Based Methods:
A modern, scalable technique where lipids in ethanol and aqueous drug solutions are mixed under controlled laminar flow.
Enables precise control over liposome size, polydispersity, and reproducibility.
Increasingly used in GMP manufacturing.
5. Extrusion and Sonication (Size Reduction):
MLVs can be processed through polycarbonate membranes (extrusion) or sonicated to achieve uniform nanosized vesicles.
Essential for reducing heterogeneity and improving pharmacokinetics.
6. Freeze–Thaw Cycling:
Repeated cycles of freezing and thawing improve encapsulation efficiency by disrupting and reforming the lipid bilayer.
Often combined with thin-film hydration.
Our team has hands-on experience in addressing each of these pain points. We provide consulting services for improving liposomal stability, training in advanced preparation techniques, and strategic guidance for scale-up and regulatory compliance. By tackling both laboratory and industrial challenges, we help ensure liposomal products reach their full potential in clinical and commercial use.
Niosomes
Niosomes are innovative vesicular carriers made from non-ionic surfactants and cholesterol, designed to encapsulate both hydrophilic and lipophilic drugs. Their importance in modern drug delivery lies in their ability to improve solubility, stability, and bioavailability while reducing systemic toxicity. However, if not optimized properly, niosomes may suffer from drug leakage, low encapsulation efficiency, or poor reproducibility, which can severely affect therapeutic outcomes.
One of the biggest advantages of niosomes is their cost-effectiveness and higher stability compared to liposomes, making them particularly attractive for companies seeking practical yet efficient nanocarrier systems. They are widely applied in transdermal delivery of anti-inflammatory drugs, cancer therapy, vaccine formulations, and cosmetics.
In practice, researchers often face real-world challenges when working with niosomes. Drug leakage during storage is common, but this can be mitigated by optimizing the surfactant-to-cholesterol ratio, incorporating stabilizers, or applying lyophilization techniques. Encapsulation efficiency, especially for hydrophilic drugs, may be disappointingly low, requiring fine-tuning of hydration methods and surfactant selection. At the scale-up stage, batch-to-batch variability is another frequent issue, where manual methods such as thin-film hydration are difficult to reproduce consistently. Transitioning to microfluidic or continuous production platforms can significantly improve uniformity and reproducibility. Finally, long-term stability can be affected by surfactant oxidation or aggregation, problems that can be addressed with antioxidant incorporation and optimized storage conditions.
Our consulting team specializes in troubleshooting exactly these types of problems. We guide clients in designing robust formulations, adopting industrially relevant production methods, and training staff on critical quality control steps. With the right strategies, niosomes can become a reliable and scalable solution for advanced drug delivery applications.
Several approaches are commonly used to prepare niosomes, each with specific advantages depending on the type of drug and the desired vesicle characteristics:
1. Thin Film Hydration (Conventional Method):
Non-ionic surfactants and cholesterol are dissolved in an organic solvent, which is then evaporated to form a thin lipid film.
Hydration with an aqueous buffer results in multilamellar vesicles (MLVs), which can be further downsized by extrusion or sonication.
2. Reverse Phase Evaporation:
Surfactants dissolved in organic solvents are emulsified with an aqueous phase followed by solvent removal.
Produces large unilamellar vesicles (LUVs) with high encapsulation efficiency for hydrophilic drugs.
3. Ether or Ethanol Injection:
A solution of surfactants in a volatile organic solvent (ether/ethanol) is rapidly injected into an aqueous solution.
Solvent evaporation leads to the spontaneous formation of niosomes.
4. Microfluidic Mixing:
Modern technique enabling precise control over vesicle size, uniformity, and reproducibility.
Highly scalable and increasingly used in industrial applications.
5. Sonication and Extrusion:
Applied to reduce particle size and polydispersity, leading to nanosized niosomes with improved stability.
Lipid Nanoparticles (LNPs)
Lipid nanoparticles (LNPs) represent the most advanced generation of lipid-based carriers, composed of ionizable lipids, cholesterol, phospholipids, and PEG-lipids. Their importance cannot be overstated: LNPs enabled the delivery of mRNA vaccines during the COVID-19 pandemic and are now revolutionizing gene therapy, siRNA delivery, and cancer immunotherapy. Unlike conventional carriers, LNPs are specifically engineered to encapsulate and protect fragile nucleic acids, ensuring that they reach target cells effectively. Without careful design, however, nucleic acids degrade rapidly, lose activity, or trigger unwanted immune responses.
The main advantage of LNPs is their high encapsulation efficiency and proven clinical success. They offer a safer alternative to viral vectors, with the flexibility to adjust lipid composition for tissue-specific targeting. Beyond vaccines, LNPs are opening doors for personalized medicine and rare disease therapies, where precision delivery is critical.
Yet, researchers and manufacturers often encounter serious challenges with LNPs. A major problem is stability and cold-chain dependency, as many formulations require ultra-low temperatures for storage, which complicates logistics and increases costs. This can be improved by engineering more stable lipid structures or developing lyophilized formulations. Batch-to-batch variability and reproducibility during lab preparation is another common obstacle, especially when using manual mixing methods. Moving to microfluidic systems and automated GMP-compliant platforms significantly enhances consistency.
Additionally, off-target delivery and unwanted immune activation remain major concerns, which can be addressed through careful optimization of ionizable lipids, PEGylation strategies, and surface modifications. From an industrial perspective, scaling up production while maintaining encapsulation efficiency is a recurring challenge; transitioning from bench-scale mixers to industrial continuous systems is often essential for commercialization.
Lipid nanoparticles are typically composed of ionizable or cationic lipids, helper phospholipids, cholesterol, and PEG-lipids. Their preparation methods are designed to ensure efficient encapsulation of nucleic acids (e.g., siRNA, mRNA) or small molecules while maintaining particle stability and uniformity.
1. Microfluidic Mixing (Gold Standard):
The most widely used method for clinical and industrial applications.
Lipids dissolved in ethanol are rapidly mixed with an aqueous solution of nucleic acids under controlled microfluidic flow.
Results in uniform nanoparticles (50–100 nm) with high encapsulation efficiency.
Scalable and highly reproducible, making it the preferred technique for GMP manufacturing.
2. Ethanol Injection:
Lipid solution in ethanol is injected into an aqueous phase containing the therapeutic payload.
Rapid self-assembly of lipids into nanoparticles occurs.
Simple and cost-effective but offers less control over particle size and polydispersity compared to microfluidics.
3. T-Mixer or Flash Nanoprecipitation:
A variant of controlled mixing using turbulent flow or staggered herringbone micromixers.
Allows precise particle size tuning and is adaptable to continuous manufacturing.
4. High-Pressure Homogenization:
Lipids and payload are pre-emulsified and then homogenized at high pressure.
Produces nanosized lipid particles with relatively uniform distribution.
More commonly applied to solid lipid nanoparticles but can be adapted for LNPs.
5. Solvent Evaporation or Dialysis:
Lipids dissolved in organic solvents are mixed with the aqueous phase and then subjected to solvent removal (by evaporation or dialysis).
Less common for industrial production due to low encapsulation efficiency and scalability challenges.
Our consulting and training programs are designed to help companies overcome these exact bottlenecks. We support clients in formulation optimization, troubleshooting encapsulation efficiency, improving stability for real-world conditions, and preparing scalable manufacturing strategies. By combining scientific expertise with practical solutions, we enable organizations to accelerate the development of LNP-based therapeutics from lab to market.
Genetic Engineering
Cloning
Cloning involves molecular cloning (Gene Cloning), which is the process of generating identical copies of a specific DNA sequence (a gene) by inserting it into a host organism, like a bacterium, which then replicates it.
Moreover, it consists of cell line cloning, which is the process of isolating a single cell and allowing it to proliferate in culture to create a population of genetically identical cells, known as a clonal cell line. This application is achieved through various techniques, with the most common being recombinant DNA technology, polymerase chain reaction (PCR), and single-cell dilution cultures. This procedure’s ability to create genetically identical copies has revolutionized biological research and medicine.
The most important application of DNA cloning is used in biopharmaceuticals, gene therapy, and gene analysis.
Cloning procedures involve creating multiple identical copies of a specific DNA fragment, often a gene, using a host organism (mammalian, eukaryotic, and prokaryotic). DNA cloning is the process of making multiple, identical copies of a particular piece of DNA. In a typical DNA cloning procedure, the gene or other DNA fragment of interest is first inserted into a circular piece of DNA called a plasmid. The insertion is done using enzymes that “cut and paste” DNA, and it produces a molecule of recombinant DNA, or DNA assembled out of fragments from multiple sources. This allows for the amplification of the gene and, in some cases, its expression to produce a protein.
Cloning technology faces significant practical challenges, including low-efficiency ligation. That can be overcome by optimizing the insert and using high-concentration T4 DNA Ligase. Restriction enzyme issues that to solve this problem, performing double digestion and optimizing digestion time & conditions are too effective. No colonies or too many empty colonies can be solved by using controls and antibiotic selection & blue-white screening are too effective. PCR-related problems can be solved by using a high-fidelity polymerase and designing effective primers. Toxic inserts can be solved by using tightly regulated expression vectors and cloning at lower temperatures.
Our team offers hands-on training and consulting to troubleshoot cloning experiments, optimize digestion time, and ensure optimal insert orientation. By addressing these challenges, we help researchers and companies achieve reliable, reproducible results efficiently.
Molecular Methods
Molecular biology techniques are common methods used in biology, which generally involve the manipulation and analysis of DNA, RNA, protein, and lipid.
Polymerase chain reaction (PCR)
This is one of the most important techniques used in molecular biology and is basically used to copy DNA. PCR allows a single DNA sequence to be amplified into millions of DNA molecules. PCR can also be used to introduce mutations within the DNA or introduce special restriction enzyme sites. In addition, PCR is used to determine whether a certain DNA fragment exists in a cDNA library. Different types of PCR include reverse transcription PCR (RT-PCR) for amplification of RNA and quantitative PCR (QPCR) to measure the amount of RNA or DNA present.
This procedure has practical applications in forensics, genetic testing, and diagnostics. PCR can also be used to test for a bacterium or DNA virus in a patient’s body: if the pathogen is present, it may be possible to amplify regions of its DNA from a blood or tissue sample.
PCR procedure requires a DNA polymerase enzyme, like DNA replication in an organism that makes new strands of DNA, using existing strands as templates. Two primers are used in each PCR reaction, and they are designed so that they flank the target region (the region that should be copied). The primers bind to the template by complementary base pairing. When the primers are bound to the template, they can be extended by the polymerase, and the region that lies between them will get copied. Steps of PCR consist of Denaturation for separating the double-stranded DNA template strand to the point where the strands start denaturing and the hydrogen bonds are broken between the nucleotide base pairs. Annealing for attachment of the forward and reverse primers to each of the single-stranded DNA template strands. The DNA polymerase is also stable enough to now bind to the primer DNA sequence. Extension is done for the synthesis and elongation of the new target DNA strand accurately and rapidly by DNA polymerase. Final Extension is done to fill in any protruding ends of the newly synthesized strands.
There are many common PCR challenges, such as no amplification that for overcoming this problem, template, primer, Mg²⁺ concentration, and cycling conditions should be checked. Non-specific amplification (multiple bands or smearing) can be overcome by increasing annealing temperature (try a gradient), switching to a hot-start polymerase, reducing primer concentration to 0.2 µM, and using Touchdown PCR. Primer-dimer formation can be overcome by using a hot-start polymerase, reducing primer concentration, and redesigning primers if the problem persists. Low yield (faint band) for overcoming this, increasing the cycle number slightly, increasing the template amount, optimizing annealing temperature (try a gradient), and ensuring extension time is sufficient. False positives in the no-template control (NTC) can be overcome by decontaminating everything and making new aliquots of all reagents.
By understanding these common challenges and their solutions, you can move from frustration to successful and reproducible PCR results. In this line, PCR reaction remains a reliable and versatile method for nucleotide analysis, performing and investigating cloning, and producing high-quality results. For expert consulting or hands-on training in PCR, reach out to our team today.
DNA extraction
DNA extraction method is one of the important steps in the PCR process. DNA samples from different sources are needed to perform a PCR reaction. Various tissues, including blood, body fluids, direct Fine needle aspiration cytology (FNAC) aspirate, formalin-fixed paraffin-embedded tissues, frozen tissue section, etc., can be used for DNA extraction. DNA extraction involves lysing the cells and solubilizing DNA, which is followed by chemical or enzymatic methods to remove macromolecules, lipids, RNA, or proteins. So, the DNA extraction is used to obtain purified DNA by using physical and/or chemical methods from a sample, separating DNA from cell membranes, proteins, and other cellular components. Different procedures for DNA extraction, involving manual and commercially available kits. DNA extraction techniques include organic extraction (phenol–chloroform method), nonorganic method (salting out and proteinase K treatment), and adsorption method (silica–gel membrane).
The general procedure follows three core steps consisting of Lysis for breaking open cells to release DNA, Precipitation for separating DNA from other cellular components (proteins, RNA, lipids) and isolating it, and Purification/Washing for removing salts and other contaminants to get a clean DNA pellet.
The most common challenges consist of low yield of DNA (not enough DNA) that can be overcome by increasing the starting material, optimizing Lysis, and preventing nuclease activity.
Poor Quality/Purity (dirty DNA) is another problem in which the DNA is presented with contamination such as protein, RNA, salt or ethanol, and polysaccharide or humic acid. These contaminants inhibit downstream applications like PCR, restriction digestion, or sequencing. For overcoming this problem, proteinase K, RNase A, column-based kits, and high-salt precipitation are used.
By understanding the biochemical principles behind each step, you can effectively diagnose and solve almost any DNA extraction problem. For expert consulting or hands-on training in DNA extraction, reach out to our team today.
Real-Time PCR
Real-time PCR, also known as quantitative PCR (qPCR), is a molecular biology technique that monitors the amplification of a DNA or RNA sequence during the polymerase chain reaction (PCR) process in real-time, as opposed to at the end of the reaction like in conventional PCR. This allows for the quantification of nucleic acids, making it a valuable tool for various applications. There are important differences between the usual PCR and Real-time PCR.
In real-time PCR, the amplification of DNA or RNA is monitored during each cycle of the PCR process using fluorescent signals. Real-time monitoring enables the quantification of the starting amount of the target nucleic acid, unlike conventional PCR, which only indicates presence or absence. Three general methods are used for the quantitative detection, such as Hydrolysis probes (TaqMan, Beacons, Scorpions), Hybridization probes (Light Cycler), and DNA-binding agents (SYBR Green).
Important agents in Real-time PCR is related to Fluorescent Detection: Real-time PCR utilizes fluorescent dyes or probes that bind to the amplified DNA, generating a fluorescent signal that is proportional to the amount of DNA present. Cycle Threshold (Ct): The point at which the fluorescence signal crosses a threshold (Ct value) is used to determine the initial amount of target DNA. A lower Ct value indicates a higher starting concentration of the target. Data Analysis: The fluorescence data collected during each PCR cycle are used to generate amplification curves, allowing for quantification of the target DNA.
The applications of Real-time PCR consist of gene expression analysis, pathogen detection, genetic testing, food safety, and research studies.
Real-time PCR procedure begins with the meticulous preparation of a reaction mix containing the template DNA (or reverse-transcribed cDNA for RT-qPCR), sequence-specific forward and reverse primers, a fluorescent reporter (typically either a DNA-binding dye like SYBR Green or a sequence-specific fluorescent probe, such as a TaqMan probe), nucleotides (dNTPs), a buffer with magnesium ions, and a thermostable DNA polymerase. This mixture is aliquoted into individual wells of a specialized optical plate or tube, which is then sealed and placed into the thermal cycler of the real-time PCR instrument. The instrument then executes a programmed series of temperature cycles—initial denaturation, followed by repeated cycles of denaturation, annealing, and extension—while simultaneously exciting the fluorescent dyes with a light source and measuring the intensity of the emitted fluorescence at the end of each cycle. As the target sequence is exponentially amplified, the accumulating product binds the fluorescent dye, causing a measurable increase in fluorescence signal that is directly proportional to the amount of amplicon generated. This data is collected in real-time by the instrument’s software, which subsequently plots fluorescence against cycle number to generate an amplification curve for each sample, allowing for the precise quantification of the initial target amount through the determination of the cycle threshold (Ct) value, where the fluorescence exceeds a background level.
Real-Time PCR is a powerful technique, but its sensitivity makes it prone to issues. Problems generally fall into a few categories: amplification issues, signal issues, data quality issues, and contamination. Amplification issues related to No Ct, Late Ct, or failed reactions. This is the most common problem in Real-Time PCR that the reaction either doesn’t amplify at all or amplifies very inefficiently. To solve this problem, the quality and degradation of the template and presentation of inhibitors such as phenol, heparin, ethanol, and salts should be checked. Moreover, primer or probe issues, which consist of wrong design, degradation, or incorrect concentration, can be solved by checking BLAST sequences to ensure the specificity and comparing the concentrations of the primer can be useful. Signal & Fluorescence issues are other problems related to the detection of the fluorescent signal. Low fluorescence signal is an important problem that creates a weak signal in even positive controls and shows high background noise. To solve this problem, keeping probes in the dark (use amber tubes) and avoiding repeated freeze-thaw cycles are highly recommended. Data quality & reproducibility issues with the reaction runs, in which the data is inconsistent or unreliable, are related to pipetting error and template inhomogeneity. Using quality pipettes, calibrating them, and mixing the template thoroughly by vortexing and spinning down all samples and master mix before use are effective ways.
By following this structured approach, investigating general troubleshooting, you can systematically diagnose and resolve most qPCR challenges. In this line, our team offers expert consulting or hands-on training in Real-Time RCR.
RNA Extraction
RNA isolation is an important step in analysing gene expression, which is a common application of RT-qPCR. Gene expression requires RNA, which is reverse transcribed into cDNA, and the cDNA in turn becomes the template for the qPCR amplification in an experiment. Hence, the first step in gene expression analysis is RNA isolation.
There are four common RNA extraction methods, which consist of Organic (Phenol-Chloroform) Extraction (e.g., TRIzol®, TRI Reagent®). Spin Column (Silica Membrane) Based Purification, Magnetic Bead-Based Purification, and Automated Extraction Systems.
In essence, RNA extraction procedure is a careful balancing act consisting of breaking open cells and tissue, inactivating RNases to prevent degradation, separating RNA from DNA, proteins, and other cellular components, washing away all contaminants, eluting the pure, intact RNA in a stable solution, and verifying its quality and quantity before use.
On the other hand, the disadvantages and limitations of RNA extraction are related to rigorous RNase inhibition, RNA instability, and co-purification of inhibitors.
RNA Extraction method is vital for Gene expression analysis, a diagnosis of RNA viruses, Research and discovery of novel RNA species, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), functional genomics, which provides a dynamic view of cellular activity, and Next-Generation Sequencing (NGS) for RNA-Seq.
The most common challenges are related to RNase contamination, RNA degradation, genomic DNA (gDNA) contamination, and low yield.
As RNases are ubiquitous and extremely robust, they don’t denature by autoclaving and can renature after heating. Using RNase-free reagents, consumables, and equipment, wearing gloves and changing them frequently, and using Diethyl pyrocarbonate (DEPC) to inactivate RNases, are highly recommended. The result of RNase activity leads to smeared and degraded RNA bands on an electrophoresis gel instead of sharp ribosomal RNA bands. So, to solve this issue, checking the quality of RNA by running it on a gel and using the correct lysis buffer-to-sample ratio to ensure immediate inactivation of RNases is necessary.
As DNA contamination can co-precipitate with RNA, especially if using silica-column methods without a DNase step. This causes false positives in qPCR. In this line, the gold standard is performing an on-column DNase I digestion step.
Furthermore, low yield during RNA extraction can be modified by starting with more sample and ensuring the tissue is thoroughly homogenized to liberate all RNA.
Mastering this technique is a cornerstone of modern molecular biology and genetics research. So, our team has hands-on experience in addressing each of these pain points. We provide consulting services for improving RNA stability and purity, training in advanced preparation techniques, and strategic guidance for scale-up and regulatory compliance. By tackling laboratory challenges, we can ensure RNA extraction products reach their full potential for other molecular techniques.
Gel Agarose Electrophoresis
Gel agarose electrophoresis is a fundamental laboratory technique used to separate DNA or RNA fragments based on size. It provides a simple yet powerful method for analyzing nucleic acids, verifying PCR products, and checking sample integrity. Its importance lies in providing reliable information on nucleic acid quality and fragment size; without proper execution, results can be smeared, faint, or misleading, impacting downstream experiments.
The advantages of agarose gel electrophoresis include its simplicity, cost-effectiveness, and rapid visualization of nucleic acids. It allows researchers to quickly assess the success of amplification or restriction digestion and is widely used in molecular biology, genetics, and diagnostics.
Despite its simplicity, laboratories often face practical challenges. Uneven gel polymerization or improper agarose concentration can cause distorted bands, while sample overloading may lead to smearing. Poor quality or degraded samples can reduce signal intensity. These issues can be addressed by optimizing gel concentration, using high-quality buffers, controlling sample amounts, and applying standardized running conditions. Additionally, safety concerns related to ethidium bromide or other dyes require proper handling and disposal protocols.
Our team offers hands-on training and consulting to troubleshoot agarose electrophoresis experiments, optimize gel and buffer compositions, and implement safer staining techniques. By addressing these challenges, we help researchers and companies achieve reliable, reproducible results efficiently.
Overall, agarose gel electrophoresis remains an essential tool for nucleic acid analysis, and expert guidance can maximize both accuracy and efficiency in laboratory workflows. For practical training or consulting in agarose gel electrophoresis, contact our team today.
SDS-PAGE Electrophoresis
SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) is a widely used technique to separate proteins based on their molecular weight. By denaturing proteins and imparting uniform negative charge, SDS-PAGE enables precise protein profiling, purity assessment, and verification of protein expression. Its importance is critical: incorrect gel preparation or running conditions can lead to distorted bands, incomplete separation, or inaccurate molecular weight estimation.
The advantages of SDS-PAGE include high resolution, reproducibility, and adaptability to downstream applications such as Western blotting or protein quantification. It is a cornerstone technique in proteomics, drug discovery, and biotechnology research.
Laboratories often encounter several challenges. Incomplete protein denaturation or sample aggregation can affect separation quality. Gel polymerization errors or improper acrylamide percentage can distort migration patterns. Additionally, overheating during electrophoresis may cause band smearing, while low-quality reagents reduce resolution. These problems can be solved by using optimized sample preparation protocols, precise gel casting techniques, and controlled electrophoresis conditions. Our consulting services guide labs in selecting the correct gel percentage for protein sizes, preparing high-quality reagents, and troubleshooting experimental anomalies.
SDS-PAGE remains a reliable and versatile method for protein analysis, and proper guidance ensures reproducible, high-quality results. For expert consulting or hands-on training in SDS-PAGE electrophoresis, reach out to our team today.
Western Blotting
Western blotting is a powerful analytical technique used to detect specific proteins in complex samples. By combining SDS-PAGE separation with antibody-based detection, it provides qualitative and quantitative insights into protein expression, post-translational modifications, and biomarker validation. Its importance is paramount: improper transfer, antibody selection, or blocking can produce weak or non-specific signals, compromising experimental conclusions.
Western blotting offers significant advantages over other protein detection methods, including high specificity, versatility in sample type, and compatibility with multiple detection systems (chemiluminescence, fluorescence, or colorimetric). Applications range from basic research in cell biology to clinical diagnostics, drug development, and biomarker discovery.
Despite its widespread use, real-world challenges are common. Inefficient protein transfer can occur due to incorrect membrane choice or running conditions. Non-specific antibody binding or high background signal often results from insufficient blocking or suboptimal antibody concentrations. Reproducibility issues may arise from inconsistent sample loading or variable gel quality. These can be addressed through careful selection of membranes and antibodies, standardized blocking protocols, and optimized transfer and detection conditions. Our team provides specialized consulting and hands-on workshops to troubleshoot these issues, improve signal quality, and ensure reproducible results.
Western blotting continues to be an indispensable tool for protein analysis, and professional guidance can significantly enhance both accuracy and reliability. For training or consulting in Western blotting, our expert team is ready to support your laboratory’s needs.
Cell processing and functional testing
Stem Cells Isolation
Stem cell isolation is a fundamental step in regenerative medicine and cell-based therapies, as it provides researchers with the raw biological material needed to study developmental biology, model diseases, and design therapeutic strategies. Stem cells can be derived from various sources, including bone marrow, adipose tissue, peripheral blood, umbilical cord blood, and embryonic or induced pluripotent stem cells (iPSCs). The choice of source depends on accessibility, ethical considerations, and intended applications. Isolation techniques aim to separate stem cells from a heterogeneous population of cells while preserving their viability, multipotency, and proliferative capacity.
Several strategies have been developed to achieve efficient stem cell isolation. Density gradient centrifugation is commonly used for separating mononuclear cells, while immunomagnetic bead sorting and fluorescence-activated cell sorting (FACS) allow for highly specific selection based on surface markers such as CD34 for hematopoietic stem cells or CD105/CD73/CD90 for mesenchymal stem cells. Advances in microfluidics and label-free methods, such as size-based filtration or dielectrophoresis, are providing new opportunities for gentle and scalable isolation without the need for extensive labeling. Each method has trade-offs between yield, purity, speed, and cost, and the selection of technique often depends on the downstream application—whether for research, preclinical studies, or clinical therapy.
Despite significant progress, stem cell isolation faces important challenges. One major limitation is heterogeneity—stem cell populations are rarely homogeneous, and isolating pure subsets while maintaining functionality is difficult. Marker-based selection can be problematic since no single marker is universally specific for stem cell populations, leading to contamination with other cell types. The process itself can induce stress or alter cell behavior, reducing viability or differentiation potential. Scaling up isolation methods for clinical-grade applications presents further difficulties, as techniques that work in the laboratory may not be suitable for large-scale or GMP-compliant production. Ethical and regulatory considerations, particularly for embryonic stem cells, also continue to shape how and where certain isolation strategies can be applied.
The workflow of stem cell isolation usually begins with tissue collection, followed by mechanical or enzymatic dissociation to release cells. The resulting suspension is then subjected to enrichment and purification steps such as density gradients, magnetic sorting, or flow cytometry. Isolated cells are subsequently characterized for viability, phenotype, and stemness markers before being expanded in culture or used directly in experiments. In clinical contexts, additional steps such as sterility testing, cryopreservation, and quality control are integrated to ensure safety and reproducibility.
By improving efficiency, reducing stress on cells, and developing scalable protocols, stem cell isolation continues to evolve as a cornerstone of regenerative medicine. The refinement of isolation methods not only enhances research reproducibility but also paves the way for safe and effective therapeutic applications, bringing stem cell–based treatments closer to routine clinical practice.
Flow Cytometry
Flow cytometry is one of the most powerful and versatile tools in modern biomedical research, offering the ability to analyze millions of individual cells in suspension within minutes. Its importance lies in providing high-throughput, multiparametric data that cannot be achieved with conventional techniques such as microscopy. By combining speed, precision, and statistical robustness, flow cytometry has become essential not only in basic science but also in clinical diagnostics and biopharmaceutical development. For example, immunophenotyping of T-cells, B-cells, and NK cells has transformed immunology research, while the routine monitoring of CD4+ T-cells in HIV patients demonstrates its clinical impact. The technology is equally valuable for cell cycle analysis, apoptosis detection, stem cell characterization, and high-throughput drug screening, making it a true workhorse across both academic and industrial laboratories.
Despite its tremendous potential, flow cytometry comes with notable challenges. Instruments are expensive and require highly trained operators to design experiments, optimize staining panels, and interpret complex datasets. Multiparametric analysis often introduces issues such as spectral overlap between fluorophores, which necessitates careful compensation and calibration. Sample preparation is another critical step, as improper handling may lead to cell clumping, artifacts, or even loss of sensitive populations. Moreover, variability between instruments and laboratories can complicate data reproducibility, making standardization a key concern for regulatory acceptance and translational applications.
The process of flow cytometry follows a series of well-established steps that together ensure reliable results. Cells are first harvested and suspended in a suitable buffer, followed by staining with fluorochrome-conjugated antibodies or fluorescent dyes that target specific surface or intracellular markers. Once prepared, the suspension is introduced into the flow cytometer, where each cell passes individually through a focused laser beam. Light scattering provides information on cell size and internal complexity, while emitted fluorescence signals are collected to reveal marker expression. With appropriate software, researchers apply gating strategies to identify distinct subpopulations, quantify cell frequencies, and extract meaningful biological insights. To ensure data quality, instruments are routinely calibrated with fluorescent beads and strict quality-control protocols.
By addressing the challenges of cost, complexity, and standardization, laboratories and companies can fully harness the potential of flow cytometry. Its ability to simultaneously capture structural, functional, and molecular features at the single-cell level continues to drive breakthroughs in cancer immunology, stem cell research, and therapeutic development. As innovations such as spectral cytometry and automated analysis expand its capabilities, flow cytometry remains a cornerstone technology at the intersection of research, clinical practice, and industrial biomedicine.
Stem Cell Differentiation Assay Methods
Stem cell differentiation assays are essential techniques used to evaluate the potential of stem cells to develop into specific cell types, such as osteocytes, adipocytes, or neurons. These assays are crucial in regenerative medicine, drug discovery, and tissue engineering because they confirm stem cell potency and guide experimental and therapeutic applications.
The main advantages of these assays include providing functional insights into stem cell biology, validating stem cell lines, and optimizing culture conditions for reproducible results. They are widely applied in research, preclinical studies, and industrial stem cell-based products.
In practice, laboratories often face challenges such as inconsistent differentiation efficiency, cell contamination, and batch-to-batch variability. Improper media composition, suboptimal induction protocols, or incorrect timing can lead to incomplete differentiation or heterogeneous populations. These issues are addressed by careful selection of differentiation factors, strict aseptic techniques, and standardized induction protocols. Our consulting and training services guide teams in designing reliable differentiation assays, troubleshooting variability, and ensuring reproducible results.
Stem cell differentiation assays remain indispensable for verifying stem cell functionality. For hands-on training or specialized consulting in stem cell differentiation, contact our expert team to optimize your workflows.
Cell Culture
Stem cell differentiation assays are essential techniques used to evaluate the potential of stem cells to develop into specific cell types, such as osteocytes, adipocytes, or neurons. These assays are crucial in regenerative medicine, drug discovery, and tissue engineering because they confirm stem cell potency and guide experimental and therapeutic applications.
The main advantages of these assays include providing functional insights into stem cell biology, validating stem cell lines, and optimizing culture conditions for reproducible results. They are widely applied in research, preclinical studies, and industrial stem cell-based products.
In practice, laboratories often face challenges such as inconsistent differentiation efficiency, cell contamination, and batch-to-batch variability. Improper media composition, suboptimal induction protocols, or incorrect timing can lead to incomplete differentiation or heterogeneous populations. These issues are addressed by careful selection of differentiation factors, strict aseptic techniques, and standardized induction protocols. Our consulting and training services guide teams in designing reliable differentiation assays, troubleshooting variability, and ensuring reproducible results.
Stem cell differentiation assays remain indispensable for verifying stem cell functionality. For hands-on training or specialized consulting in stem cell differentiation, contact our expert team to optimize your workflows.
Cell proliferation
Cell culture and expansion are foundational techniques for generating sufficient numbers of healthy cells for research, therapy, or industrial applications. Maintaining optimal growth conditions is critical for cell viability, functionality, and consistency in downstream applications.
The advantages of well-executed cell culture include controlled growth, scalability, and the ability to manipulate cell conditions for experimental or therapeutic purposes. Applications range from basic research and drug screening to cell therapy and regenerative medicine.
Common real-world challenges include contamination (bacterial, fungal, or mycoplasma), cell senescence, and variable growth rates. Using suboptimal media, improper incubator conditions, or inconsistent handling can compromise cell health. Solutions include implementing strict aseptic protocols, using optimized media formulations, monitoring cell morphology, and training staff in best practices. Our consulting and training services help labs improve culture reliability, scale-up efficiency, and quality control processes.
Cell culture and expansion are the backbone of all cellular research and therapy. For expert consulting or hands-on workshops in cell culture, connect with our team to ensure robust and reproducible outcomes.
Cell Counting
Cell counting is a fundamental technique used to determine cell concentration and viability, providing critical information for experimental planning, seeding cultures, and quality control. Accurate cell counts are essential for reproducible results in research and production.
The advantages include precise dosing, monitoring proliferation, and optimizing resource use. This technique is universally applied in drug screening, stem cell studies, and biomanufacturing.
Laboratories often face challenges such as clumped or dead cells leading to inaccurate counts, manual counting errors, and instrument calibration issues. These problems can be solved through proper sample preparation, using automated counters or viability dyes, and regular equipment maintenance. Our consulting and training services help labs implement reliable counting protocols, minimize human error, and standardize quality control procedures.
Accurate cell counting is essential for reproducible research and clinical-grade cell production. For training or consulting on advanced cell counting techniques, our team provides practical guidance to optimize accuracy and efficiency.
Cell Freeze (Backup Preparation) / Defreeze
Cell freezing and thawing are critical procedures for long-term cell storage, quality assurance, and experimental reproducibility. Proper cryopreservation ensures cell viability, phenotype maintenance, and readiness for downstream applications.
The advantages include creating reliable backup stocks, maintaining genetic integrity, and enabling flexible research planning. Applications range from stem cell banking, biobanking, and regenerative medicine to routine laboratory experiments.
Common challenges include reduced viability after thawing, ice crystal formation damaging cells, and variability in recovery between batches. These issues can be addressed by using optimized cryoprotectants, controlled-rate freezing, and standardized thawing protocols. Our team provides consulting on cryopreservation strategies, hands-on training in freeze/thaw techniques, and troubleshooting to maximize post-thaw recovery and functionality.
Effective cell freezing and thawing is essential for consistent and reliable cell-based research. For expert guidance or hands-on workshops in cell cryopreservation, reach out to our experienced team today.
Cytotoxicity Assay
MTT Assay
The MTT assay is a widely used method to assess cell viability and metabolic activity. It relies on the reduction of the yellow MTT reagent into insoluble purple formazan crystals by mitochondrial enzymes in living cells. This assay is critical in drug development, cytotoxicity evaluation, and stem cell research, as it provides a quantitative measure of cell health.
One of the main advantages of the MTT assay is its simplicity, cost-effectiveness, and reproducibility, making it suitable for high-throughput screening. It is widely applied in pharmaceutical testing, biomaterial evaluation, and cancer research to assess the effects of compounds on cell proliferation.
In practice, labs often encounter challenges such as incomplete solubilization of formazan crystals, compound interference due to inherent color, and well-to-well variability. These issues can lead to inaccurate readings and misinterpretation of results. Solutions include optimizing solubilization protocols, using proper controls, standardizing cell seeding densities, and training staff on precise handling. Our team provides consulting and hands-on training to overcome these challenges, ensuring reliable and reproducible outcomes.
The MTT assay remains a cornerstone technique for cytotoxicity evaluation. For expert guidance, training, or protocol optimization in MTT assays, contact our experienced team today.
Alamar Blue Assay
The Alamar Blue assay is a sensitive, non-toxic method for assessing cell viability and metabolic activity. It uses resazurin, a blue dye, which is reduced to resorufin, a fluorescent compound, by viable cells. This allows real-time monitoring of cell proliferation and cytotoxicity without harming the cells.
Its main advantages include compatibility with high-throughput formats, non-destructiveness (enabling repeated measurements on the same sample), and high sensitivity. Alamar Blue is extensively used in drug discovery, nanomaterial evaluation, stem cell studies, and toxicity screening.
Despite its utility, laboratories may face challenges such as variable incubation times affecting signal intensity, reagent instability, and interference from colored media or test compounds. These issues can compromise accuracy if not properly addressed. Solutions include standardizing assay timing, using freshly prepared reagents, applying appropriate controls, and optimizing sample handling. Our consulting and training services help laboratories implement robust Alamar Blue protocols, troubleshoot variability, and interpret results with confidence.
The Alamar Blue assay provides a flexible, reliable approach to measuring cell viability. For hands-on training or consulting in Alamar Blue assays, reach out to our expert team to optimize your workflows.
Migration& invasive Assay
Invasive Assay
An invasion assay is a laboratory technique used to measure the ability of cells to invade through a barrier that mimics the extracellular matrix (ECM) and basement membranes in the body.
The purpose of the invasion assay is related to investigating metastasis, inflammation, and embryonic development, cancer research, drug discovery, and basic biology.
Boyden Chamber Assay (Transwell Invasion Assay) is the gold standard for this test, which uses a special insert with a porous membrane. Matrigel is an important component in this assay, which consists of a gelatinous protein mixture secreted by mouse tumor cells. Before the experiment, a thin layer of Matrigel is applied to the top of the membrane in the insert, creating a barrier that cells must actively degrade and invade through.
The procedure of the invasion assay includes placing a well containing cell culture medium that often contains a chemoattractant—a substance that lures cells to move toward it (e.g., a growth factor like FBS). In the upper chamber, cells being tested are seeded in a serum-free medium and after 24-48 hours of incubation, the chemoattractant in the lower chamber creates a concentration gradient. The cells are motivated to move downward toward the attractant. In conclusion, non-invasive cells remain on the top surface of the matrigel and invasive cells are found on the bottom surface of the membrane. By staining the cells and counting the number that have successfully invaded through to the bottom side, are done.
The entire process is fraught with potential challenges that can lead to high variability, no invasion, or false results. Some of the problems are related to Matrigel handling and coating because it gels at room temperature (22-35°C) but is liquid at 4°C. Premature gelling during handling can create an uneven, lumpy coating that is either impenetrable or inconsistently thick. For solving this problem, keeping all procedures in a cold condition and optimizing the concentration is highly recommended. The Wiping Step is the most hands-on and variable. Incomplete wiping leaves non-invasive cells behind, inflating the invasion count. Over-wiping can damage the membrane and dislodge the cells you want to count. To fix this problem, using a cotton-tipped swab moistened with PBS and checking the wiping under a microscope to ensure all non-invasive cells are removed is very important.
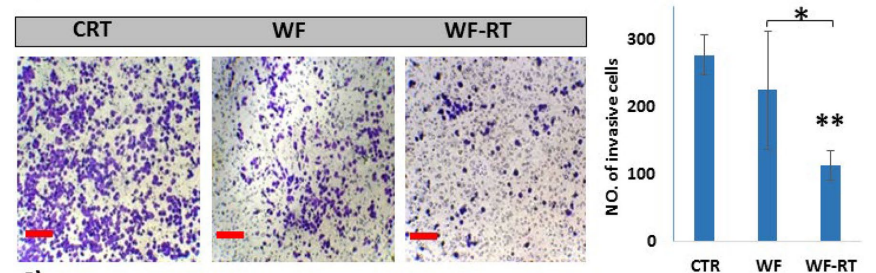
Effects of WFs on invasion BC cell line (MDA-MB 231) and in human-derived tumor spheroids. The images of the cells in the upper chamber of the Transwell assay and the graph present the percentage of cell migration. https://doi.org/10.1038/s41598-022-11023-z
The invasion assay is a cornerstone of cancer metastasis research. While its technical challenges are significant, primarily centered on the variability of Matrigel, cell health, and the manual wiping/counting steps, they are not insurmountable. By understanding these potential pitfalls and implementing the problem-solving strategies, you can generate reliable and meaningful data that accurately reflects the invasive potential of your cells. Our consulting team specializes in troubleshooting these types of problems exactly.
Scratch assay
The “Scratch” or “wound” test is a basic concept in which a monolayer of cells is observed and measured over time as the cells move to close the gap. It’s one of the oldest and most straightforward methods to study this critical biological process. The importance of the scratch assay is to investigate cell migration in many physiological and pathological conditions, such as wound healing, embryonic development, immune response, and cancer metastasis. The scratch assay is a widely used, simple, and cost-effective technique to study collective two-dimensional cell migration in vitro, as it requires minimal equipment and allows for the straightforward visualization and quantification of how fast cells close the wound, providing direct insight into processes like wound healing, metastasis, and the effects of drugs or genes on cell motility. However, its major limitation is that it only models a specific type of mechanical wounding and fails to recapitulate the complex three-dimensional microenvironment and cell-matrix interactions present in actual physiological or pathological processes.
The procedure of the scratch assay includes cell culture with a confluent (100%) monolayer that covers the entire surface. Creating the “Scratch” as the key step, in which the monolayer is scratched in a straight line to create a cell-free gap. The dish is gently washed with a buffer solution to remove the dislodged cells and debris. In this step, the treatment is added to see its effect on migration. Eventually, imaging, monitoring, and analysis are done by placing them under a microscope and taking pictures at regular intervals (e.g., every 12, 24, and 48 hours). The images are analyzed using software (like ImageJ) to measure the width of the scratch at each time point.
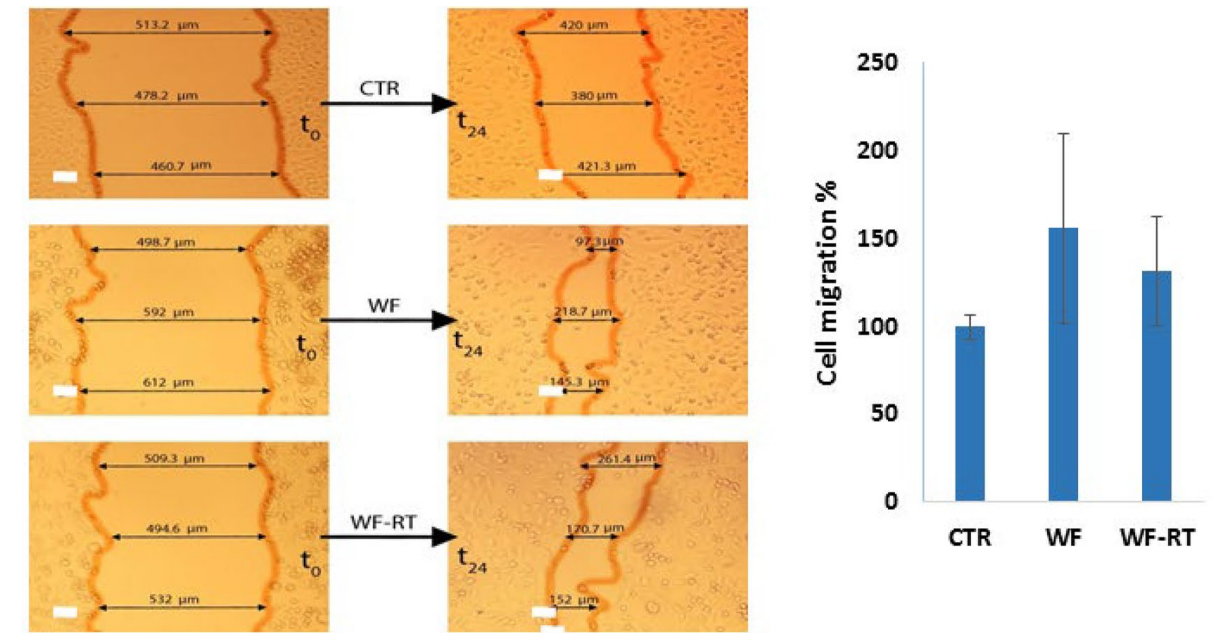
Effects of WFs on migration of BC cell line (MDA-MB 231) and in human-derived tumor spheroids. Images and graphs related to scratch assay (wound healing compared to negative control in 0 and 24 h). The graph presents the percentage of migrated cells. https://doi.org/10.1038/s41598-022-11023-z
The challenge in scratch assay can be broken down into three phases: scratch creation, post-scratch handling, and imaging/analysis.
The most important issue in scratch creation is related to inconsistent scratch width, which makes quantification unreliable. To solve this problem, practicing and repeating the technique and using a skilled guide can be useful.
The most important issue in post-scratch handling is related to cell death or detachment and proliferation vs. migration. Therefore, to decrease cell death, all washing and media changes should be done gently. To investigate migration, Mitomycin-C is used to inhibit or halt cell division without affecting migration.
The most important issue in imaging and analysis is related to subjectivity in analysis. Therefore, to remove this issue, using automated software, such as ImageJ (Fiji) and blind analysis, is highly recommended.
The scratch assay is a classic example of a technique where the art of the researcher is as important as the science. Careful attention to these challenges will lead to robust and publishable data. Hence, our consulting and training programs for scratch assay are designed to help researchers overcome these exact bottlenecks.
Tube Formation Assay
The Tube Formation Assay is used to study angiogenesis and the process of forming new blood vessels from pre-existing ones. By seeding endothelial cells on top of the Matrigel as a basement membrane extract (BME), investigating cells attach, migrate, align, and form an interconnected is explained. The resulting structures are then quantified by measuring metrics like total tube length, number of branch points, number of meshes, and total mesh area.
Advantages of the Tube formation assay include rapid and high-throughput capable, highly reproducible (When Optimized), excellent for phenotypic screening, relatively simple and inexpensive, amenable to quantification, and reduces animal Use (3Rs Principle). However, a significant limitation of this assay is its oversimplification of the complex in vivo angiogenic microenvironment, which can lead to results that may not fully predict in vivo efficacy or biological complexity.
The application of tube formation assay is confirmed to assess the pro- or anti-angiogenic effects of various compounds, drugs, gene modifications, or conditioned media from other cell types. This procedure is also used for screening potential cancer therapeutics, studying vascular biology in diseases like diabetes, and testing the efficacy of regenerative medicine approaches designed to promote vascularization in engineered tissues.
The tube formation assay procedure begins by thawing and gently pipetting a basement membrane matrix, like Matrigel, into the wells of a pre-chilled multi-well plate, which is then incubated to allow the matrix to polymerize into a solid gel. Subsequently, endothelial cells, such as Human Umbilical Vein Endothelial Cells (HUVECs), are seeded as a single-cell suspension on top of the set gel and incubated. Over a period of several hours (typically 4-18 hours), the endothelial cells will spontaneously form interconnected, mesh-like structures that mimic early capillary networks, which are finally visualized and quantified using an inverted light microscope and image analysis software to measure parameters like total tube length, number of junctions, and mesh area.
One of the important biological challenges in interpretation is that the assay doesn’t tell the whole story of angiogenesis. It measures differentiation, not proliferation. To solve this issue, complementing the tube assay with a proliferation assay (e.g., MTT, BrdU) is highly recommended.
By understanding, training, and addressing the challenges through our team, you can transform the tube formation assay from a finicky art into a robust and powerful quantitative tool for angiogenesis research.
Apoptosis Assay
Based on the central role of apoptosis, accurately detecting and quantifying apoptosis is vital in fields like cancer research, neurobiology, immunology, and drug discovery. Because it’s a multi-step process with distinct biochemical and morphological changes (cell shrinkage, membrane blebbing, DNA fragmentation, etc.), no single assay can capture every aspect. This is why the choice of assay and its interpretation are critical.
The main advantage is the ability to precisely identify and measure a specific, fundamental biological process. This breaks down into several key benefits of specificity over viability assays, mechanistic insight, high sensitivity and quantification, and versatility with multiple readouts.
There are some important applications of the apoptosis assay, including drug discovery, toxicology, and basic research.
Apoptosis assays are categorized into several groups, including assays based on Phosphatidylserine (PS) Externalization, which is one of the earliest and most reliable markers. Assays based on caspase activation that target the core apoptotic machinery and divided into immunoblotting, fluorometric, and caspase-specific antibodies. Assays based on DNA fragmentation, which is a classic hallmark of late apoptosis and consists of TUNEL assay (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) and DNA laddering. Assays based on mitochondrial changes that detect the early initiation phase of the intrinsic apoptotic pathway. Eventually, morphological assays, which are the most straightforward method used for initial confirmation, are detected by light microscopy and fluorescence microscopy with DNA-binding dyes such as Hoechst 33342 and DAPI.
The procedure of apoptosis assays is used to detect and quantify the characteristic biochemical and morphological changes that occur in programmed cell death.
The general procedure begins by inducing apoptosis in a cell population (e.g., with drugs or radiation) and preparing the cells for analysis. Depending on the specific assay chosen, the technique involves staining cells with fluorescent dyes or antibodies that target key apoptotic markers, such as the externalization of phosphatidylserine (detected by Annexin V binding), the disruption of the mitochondrial membrane potential, the activation of executioner caspases, or the fragmentation of nuclear DNA. After staining, the cells are analyzed using specialized instrumentation like flow cytometry for high-throughput, quantitative data or fluorescence microscopy for visual confirmation of cellular morphology.
The main challenge stems from the fact that apoptosis is not a single event but a dynamic multi-pathway process with distinct stages:
Early Apoptosis: Initiation phase. Key events include phosphatidylserine (PS) exposure on the outer leaflet of the cell membrane and mitochondrial membrane depolarization.
For detecting this step, Annexin V / PI is used, which is better used as a dual-color assay.
Mid Apoptosis: Execution phase. Activation of caspase enzymes.
For detecting this step, Caspase Activity is used with specific caspase substrates/inhibitors.
Late Apoptosis: Degradation phase. DNA fragmentation, membrane blebbing, and formation of apoptotic bodies. For detecting this step, TUNEL assay is used.
Secondary Necrosis: Final phase. Loss of membrane integrity, which can look like primary necrosis. Detecting membrane integrity cannot easily distinguish a late apoptotic cell. Hence, multi-parameter assays are the gold standard, such as Annexin V/PI dual staining and Propidium Iodide (PI) for solving this problem.
Our team offers hands-on training and consulting to troubleshoot and implement the solutions to apoptosis assays. By addressing these challenges, we help researchers and companies significantly increase the accuracy and reliability of their apoptosis data.
TUNEL Assay
The TUNEL assay (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) is a specialized technique for detecting DNA fragmentation in apoptotic cells. It is crucial for evaluating programmed cell death in response to drugs, radiation, or toxic compounds, and for studying apoptosis in developmental biology and cancer research.
The main advantages of TUNEL assays include high specificity for apoptosis detection, compatibility with tissue sections and cultured cells, and the ability to provide both qualitative and quantitative data. It is widely applied in toxicology, cancer research, and stem cell studies to confirm cytotoxic effects at the molecular level.
However, laboratories frequently encounter challenges such as false positives due to improper fixation or permeabilization, enzymatic reaction variability, and difficulty distinguishing apoptotic from necrotic cells. Addressing these issues requires careful optimization of fixation, permeabilization, and labeling steps, alongside rigorous controls. Our team provides consulting and hands-on training to optimize TUNEL protocols, minimize background signals, and ensure reproducible, reliable apoptosis detection.
TUNEL assays remain indispensable for studying apoptosis and cytotoxicity at the molecular level. For expert consulting or hands-on training in TUNEL assays, contact our team to enhance accuracy and reliability in your experiments.
Quality Unit (QU)
In the pharmaceutical industry, the QU is an independent organizational entity responsible for ensuring the safety, efficacy, and quality of drug products by establishing and maintaining the Pharmaceutical Quality System (PQS) and ensuring compliance with regulatory requirements, such as current Good manufacturing Practices (cGMP). The Quality Unit oversees all aspects of the product lifecycle, from development to distribution, encompassing quality assurance (QA) and quality control (QC) functions, including setting standards, testing, auditing, and investigating deviations
Establishing a strong Quality Unit in the pharmaceutical industry involves a robust quality culture, clear roles, comprehensive training, and adequate resources, while key challenges include regulatory compliance, managing complex supply chains, talent shortages, resistance to change, and keeping up with evolving GMPs and technological advancements. Successfully building and maintaining a Quality Unit is crucial for patient safety, operational consistency, and meeting stringent regulatory expectations.
Key Functions of the Quality Unit
• Establishes the Pharmaceutical Quality System: The QU creates and implements the comprehensive PQS framework, which is based on international guidelines and regulatory standards like those from the FDA and ICH.
• Ensures Regulatory Compliance: It maintains adherence to all relevant national and international regulations, such as CGMPs, and ensures that all activities comply with the PQS and marketing authorizations.
• Manages Quality Control (QC) Activities: This includes performing laboratory tests on incoming components, in-process materials, packaging, and finished products using validated methods to ensure they meet specifications.
• Conducts Quality Assurance (QA) Functions: The QU audits compliance to policies and procedures, approves or rejects all GMP-related documents and materials, and investigates any non-conforming products or processes.
• Oversees Product Lifecycle: The QU’s responsibilities extend across the entire product lifecycle, including product development, manufacturing, packaging, labeling, and distribution.
• Promotes Continuous Improvement: The unit fosters a culture of continuous improvement by assessing quality systems, identifying flaws, and planning for corrective actions to enhance product quality and process capability.
• Maintains Independence: To ensure objective quality decisions, the Quality Unit must remain independent from production and other functional areas
Key Aspects of Quality Unit Establishment
Quality Culture:
Fostering a mindset where quality is ingrained in every aspect of the organization, requiring a cultural shift and consistent executive management support.
Clear Roles and Responsibilities:
Defining specific roles and responsibilities within the Quality Unit to ensure proper oversight and decision-making processes.
Robust Data Governance:
Implementing strong data governance systems to ensure data integrity and support decision-making.
Employee Training:
Providing adequate training, communication, and continuous education programs to empower employees with the necessary knowledge and skills.
Resource Allocation:
Allocating sufficient resources, including technology and infrastructure, to effectively support quality initiatives.
Quality by Design (QbD):
Adopting innovative and systematic approaches like QbD, which emphasizes understanding product and process characteristics from the outset to build quality in, rather than relying solely on testing.
Common Challenges
Regulatory Compliance: Navigating complex and ever-changing regulatory requirements and evolving GMP standards.
Supply Chain Management: Overcoming deficiencies, including late deliveries, staffing challenges, and unavailable Active Pharmaceutical Ingredients (APIs).
Talent Shortage: A general shortage of qualified personnel in the pharmaceutical sector.
Resistance to Change:
Overcoming internal resistance to new quality systems and a quality culture.
Resource Limitations: Financial challenges in allocating sufficient resources for quality initiatives, especially for smaller companies.
Technological Advancement: Keeping up with the pace of new digital technologies and the complexities of scaling up from laboratory to commercial production.
Overly Stringent Specifications: The risk of overly strict specifications leading to unnecessary recalls and drug shortages.
Data Security: Addressing growing concerns around data security and cyber threats.
Quality Metrics for Drug Manufacturing
• Chemistry, Manufacturing, and Controls Development and Readiness Pilot (CDRP) Program
• Search for Pharmaceutical Quality Documents
• Current Good Manufacturing Practice (CGMP) Regulations
• CDER Quality Management Maturity
• Q&A on CGMP Requirements
• Inspection | Enforcement Resources
• Questions and Answers on Quality-Related Controlled Correspondence
What are Quality Metrics (QM)?
QMs are an objective way to measure, evaluate, and monitor the product and process lifecycle. Quality metrics data may lead to higher levels of safety, efficacy, delivery, and performance.
Quality metrics are used throughout the drug and biological product industry to monitor processes and drive continuous improvement efforts in manufacturing. Effective use of quality metrics is one characteristic of robust site Quality Management Maturity (QMM).
Why are Quality Metrics Important?
The minimum standard for ensuring that a manufacturer’s products are safe, effective and of sufficient quality is compliance with current good manufacturing practice (CGMP) requirements. CGMP compliance alone, however, does not indicate whether a manufacturer is investing in improvements and striving for sustainable compliance, which is the state of having consistent control over manufacturing performance and quality. Sustainable compliance is difficult to achieve without a focus on continual improvement.
An effective Pharmaceutical Quality System (PQS) ensures both sustainable compliance and supply chain robustness. Quality metrics can contribute to a manufacturer’s ability to develop an effective PQS because these data provide insight into manufacturing performance and enable the identification of opportunities for updates and innovation to manufacturing practices. Quality metrics also play an important role in supplier selection and can inform the oversight of contract activities and material suppliers, as well as help determine appropriate monitoring activities to minimize supply chain disruptions. Quality metrics data from establishments can also have utility to the FDA:
Assist in developing compliance and inspection policies and practices.
Improve prediction and possibly mitigation of future drug shortages, while encouraging the pharmaceutical industry to implement innovative quality management systems for manufacturing.
Enhance FDA’s risk-based inspection scheduling, reducing the frequency and/or length of routine surveillance inspections for establishments with quality metrics that suggest sustainable compliance. Provide ongoing insight into an establishment’s operations between inspections.
How Can Quality Metrics be used at FDA?
As part of FDA’s ongoing adoption of risk-based regulatory approaches, the agency is proposing to develop and implement a Quality Metrics Reporting Program to support its quality surveillance activities. Under this program, the FDA intends to analyze the quality metrics data submitted by establishments to:
obtain a more quantitative and objective measure of manufacturing quality and reliability at an establishment; integrate the metrics and resulting analysis into FDA’s comprehensive quality surveillance program; and
apply the results of the analysis to assist in identifying products at risk for quality problems (e.g., quality-related shortages and recalls).
Background on FDA’s QM Program
In 2004, FDA issued the report “Pharmaceutical CGMPs for the 21st Century – a Risk-Based Approach”. In 2014, FDA gained additional insight when the Brookings Institute collaborated with FDA to convene an expert workshop, “Measuring Pharmaceutical Quality through Manufacturing Metrics and Risked-Based Assessment”, which provided an opportunity for pharmaceutical manufacturers, purchasers, regulators, and other stakeholders to discuss the goals, objectives, and challenges for a pharmaceutical QM program.
In July 2015, FDA issued the draft guidance Request for Quality Metrics (80 FR 44973), which described a proposed mandatory program for product-based reporting of quality metrics. Under this program, manufacturers would have submitted four primary metrics. Stakeholder comments on the guidance included concerns regarding the burden associated with collecting, formatting, and submitting data at a product level across multiple establishments; technical comments on the proposed metrics and definitions; and legal concerns regarding the proposed mandatory program. Stakeholder commenters also suggested a phased-in approach to allow learning by both industry and FDA.
In response to this feedback, FDA published a revised draft guidance in November 2016 entitled Submission of Quality Metrics Data (81 FR 85226). The 2016 guidance described an initial voluntary phase of the QM Reporting Program, with participants reporting data either by product or establishment, through an FDA submission portal. FDA removed one of the four metrics from the 2015 draft guidance and requested submission of the remaining three key metrics. This 2016 guidance also described how FDA intended to utilize the submitted data. Commenters requested a better understanding of the value and utility of the data to be submitted to FDA and how FDA would measure success of the program. Commenters also expressed a preference for a pilot program to gather industry input before implementing a widespread Quality Metrics Reporting Program.
In Federal Register notices issued in 2018, FDA announced the availability of two pilot programs, a Quality Metrics Site Visit Program (83 FR 30751) and a Quality Metrics Feedback Program (83 FR 30748) for any establishment that had a quality metrics program developed and implemented by the quality unit and used to support product and process quality improvement. Additional information and lessons learned by FDA can be found on the FDA Quality Metrics Reporting Program; Establishment of a Public Docket; Request for Comments.
In March 2022, FDA established a docket to solicit comments on changes to FDA’s previously proposed Quality Metrics Reporting Program. This notice describes considerations for refining the Quality Metrics Reporting Program based on lessons learned from two pilot programs with industry that were announced in the Federal Register in June 2018, a Site Visit Program and a Quality Metrics Feedback Program, as well as stakeholder feedback on FDA’s 2016 revised draft guidance for industry Submission of Quality Metrics Data.
Common Compliance Issues in the Pharmaceutical Industry (and How to Avoid Them)
The US FDA releases an annual report summarizing observations from inspections by industry. The pharma industry in 2018 received 3,344 observations for 390 categories of noncompliance. Almost a third of these observations (39% of them) can be divided into just ten categories, thus illustrating the common challenges of pharmaceutical compliance.
However, the results become even more apparent when you group the data into categories based on the top ten reasons.
The four-and-a-half-dozen organizations received a 483-letter relating to noncompliance for creating or following procedures or problems with record-keeping. Additionally, 322 of the pharmaceutical industries had difficulty designing and implementing adequate controls, and 137 had trouble maintaining their products.
Whenever a pharmaceutical startup or scale-up approaches market approval, it is customary to go through growing pains. A laboratory manager may discover that a batch of maintenance records was not reviewed the week before he went on vacation, for example, even though your means of doing things seem to be working. Fortunately, there is an easier way to handle an FDA inspection than simply hoping for the best.
1. Lack of Clearly Defined Procedures and SOPs
In a Standard Operating Procedure (SOP), clear steps are outlined for carrying out specific tasks in the workplace. Using an SOP simplifies communication and makes it easier to perform the necessary functions for the work to move forward. However, compliance issues tend to arise due to a lack of effective SOPs/Written Procedures.
Various issues prevent creating and using SOPs, including complicated language, lack of standardization, and inadequate training.
Related Articles: Workplace Productivity Depends on Employee’s Training and Development
2. Inadequate Maintenance Facilities
During 2018, more than 2% of the FDA observations were for inadequate cleaning, sanitizing, and maintenance. Sanitizing, maintaining, and cleaning equipment and utensils appropriately must comply with the FDA Code of Federal Regulations.
The company should clearly outline the methods for cleaning and maintaining hygienic conditions. For example:
Providing clear instructions for cleaning.
Indicating who is responsible.
Planning your cleaning schedule.
Providing the guidelines for maintaining the equipment properly.
Ensuring regular inspections and protective measures for equipment.
Additionally, it is essential to maintain cleaning activities logs and update them as often as other operational logs.
3. Not properly utilizing data
Real-time access to data allows companies to stay abreast of changes in compliance and improve performance. By doing this, an organization can minimize the effects of non-compliance effectively. Unfortunately, one of the key reasons that organizations cannot utilize the available data is outdated technology.
Compiling data from legacy systems is challenging. Most legacy systems provide incorrect data, and integrating new data is a complex process. It further keeps pharma companies from compliance reporting. Organizations often lack adequate reporting systems. To solve this problem, they use manual reporting systems, but they are susceptible to error. Additionally, the long-term costs are high. Companies should prioritize compliance needs and failing to do so could have dire consequences.
4. Inadequate laboratory control
About 4% of all FDA observations in 2018 concerned failed laboratory controls. To maintain the laboratory data, it is necessary to monitor and maintain the laboratory controls. In addition, raw data can help determine many items, from instrument calibration to employee compliance to SOP adherence- which can prevent problems from arising.
5. A lack of communication and collaboration
A SOP without clarified roles and responsibilities creates ambiguity. The employee is better able to perform their duties and remain compliant with standards if they correctly understand what is expected of them. Moreover, providing compliance training at regular intervals keeps them prepared for the unforeseen challenges they may face in their tasks. It would be best if you equip them with tools so they can communicate, collaborate, and learn new skills. For example, you can utilize a next-generation training management software to manage employee training while giving them a centralized platform to communicate and collaborate.
6. Participation among departments is low.
Developing an SOP isn’t a linear process. You must update it to keep it current. It shouldn’t be the responsibility of one department to create and maintain these documents. To ensure that the SOPs remain relevant, departments that use them need to collaborate and update them as required. In addition to encouraging feedback, employees will offer suggestions for improvements. It will help maintain a culture of quality and continuous improvement. You may need a good Change Control Software to manage changes in processes, documents, facilities, and more.
7. Faulty Product Review Records
To avoid omissions, the review and investigation processes must be clearly defined. Several ways are available for organizations to become out of compliance with CFR 211.192, which include:
Failure to perform a thorough log review
Reviewing downtime, cleaning, and clearance logs is a must
Process failures
Lab workers can’t examine their records
Lack of standardization
An operational team and the quality control unit should use a single set of standards and standard operating procedures for batch record review to prevent any misunderstandings.
It is unacceptable for a pharmaceutical company to receive a 483-letter due to its first FDA inspection. The 483 letter signifies that your organization is out of compliance, even if you have corrected the problem. The long-term costs of uncorrected quality and operational issues outweigh the cost of continuously following FDA best practices.
The causes of noncompliance with cGMP are rarely willful. Typically, 483 observations are the result of oversights. This may be the case as the laboratory manager forgot to review maintenance records after a week on vacation. An older copy of the SOP may be used by lab employees who lost the updated document. A broken workflow or collaboration may result in noncompliance issues or simply human error. Fortunately, your quality management system can help you avoid many of these issues.
Do you worry about getting an FDA 483 Form letter or a warning letter?
A Form 483 observation and a Warning Letter are not the same.
A Form 483 is a preliminary notice of potential violations observed during an FDA inspection, while a Warning Letter is a more serious, formal enforcement action taken when observations from a 483 are not adequately addressed, and it signals the potential for greater consequences. The 483 is an informal discussion tool between the FDA investigator and the company, whereas the Warning Letter is a formal, public document that requires a prompt and adequate corrective action plan.
FDA Form 483 (Notice of Inspectional Observations): A document provided by an FDA investigator at the end of an inspection, listing conditions that appear to violate the Food, Drug, and Cosmetic Act or other regulations.
• Purpose: To discuss the findings with the company and provide an opportunity for them to provide information or take corrective actions.
• Seriousness: Less severe than a Warning Letter; it is not a final determination of a violation.
• Publicity: Generally not made public, although they can be purchased.
• Requirement to Act: A company is encouraged to respond with corrective actions but is not legally obligated to act on the observations.
FDA Warning Letter: A formal, written notification from the FDA to a regulated company detailing significant violations of FDA regulations.
• Purpose: To formally state the severity of the violations and indicate the FDA’s intent to take further enforcement action if the company fails to promptly and adequately resolve the issues.
• Seriousness: A significant escalation from a 483, indicating serious compliance failures that may impact public health.
• Publicity: A public document that is posted on the FDA website.
• Requirement to Act: A company is legally required to address the concerns and make necessary changes
A warning letter is a formal, written communication from an employer to an employee/ a regulatory agency to a company, detailing a specific violation of policies, rules, or laws. Its purpose is to formally document misconduct or performance issues, clearly state expectations for improvement, and outline the consequences of not taking corrective action, such as further disciplinary measures or legal action. Warning letters serve as a record of the issue and provide a formal opportunity for the recipient to address and resolve the problem.
• Purpose: To formally address performance problems or violations of company policy that an employee needs to improve.
• Contents: Should include the specific issue, factual examples, dates, the impact of the behavior, clear steps for improvement, and potential consequences like termination.
• Goal: To give the employee a documented opportunity to correct their behavior and prevent future issues.
Key characteristics of a warning letter:
• Formality: Uses a formal tone and often requires a signature to acknowledge receipt.
• Specificity: Clearly outlines the exact issue and provides concrete examples, dates, and relevant details.
• Documentation: Creates a formal record of the problem and the steps taken to address it.
• Corrective Action: Provides clear, actionable steps for the recipient to take to resolve the issue.
• Consequences: Warns of potential repercussions if the behavior or violation is not corrected.
An FDA 483, formally the Notice of Inspectional Observations, is a document issued by the U.S. Food and Drug Administration (FDA) to companies after an inspection to detail conditions or practices that may violate the Food, Drug, and Cosmetic Act (FD&C Act) or other regulations. It serves as a formal notification from the investigator to the inspected company’s management about potential issues, but it is not a final determination of noncompliance. Companies are expected to provide a response to the observations, outlining their corrective actions to bring the facility into compliance.
According to FDA data, 3,344 observations were written to pharmaceutical firms in 2024. However, the best time to stay in compliance with cGMP for quality is before you feel the consequences of a failing inspection. The best way to ensure you don’t get a 483 is to focus on the 7 most common and likely areas and compliance issues that cause them and how you can handle them.
Challenges in pharmaceutical Quality Assurance (QA)
stringent and evolving global regulations, complex supply chains, ensuring data integrity in digital systems, attracting and retaining skilled talent, the transition from manual to digital/automated processes, communication gaps between departments, and pressure to control costs while maintaining high quality and compliance standards.
Regulatory Challenges
• Evolving Global Regulations: Pharmaceutical companies must navigate a complex and constantly changing landscape of regulations from bodies like the FDA and EMA, requiring constant updates and training for QA teams.
• Documentation: Meticulous record-keeping is essential, but managing the sheer volume of documentation and ensuring its accuracy is a significant challenge.
Process and Technology Challenges
• Complex Manufacturing Processes: The intricate nature of pharmaceutical manufacturing necessitates robust QA systems and continuous monitoring to ensure consistency and compliance across all stages.
• Data Integrity: With increasing use of electronic systems, ensuring the accuracy, security, and traceability of data is a top priority.
• Technology Integration: Adapting to and integrating new digital workflows, electronic systems, and automation can be difficult and requires significant investment in training and new infrastructure.
Operational & Human Resource Challenges
• Talent Shortage: There is a high demand for specialized QA professionals, making it difficult to attract and retain skilled individuals.
• Communication Gaps: Coordinating effectively with various departments, including production, research, and regulatory affairs, can be challenging.
• Supply Chain Management: Ensuring quality across a complex global supply chain requires strong supplier qualification and management processes.
• Cost Pressure: The need to balance high-quality standards and strict compliance with pressure to reduce costs is an ongoing challenge.
The primary challenges in pharmaceutical Quality Control (QC)
Regulatory compliance with complex, evolving global standards, data integrity issues and management, supply chain complexities, the need to keep pace with digitalization and technological advancements, maintaining a strong quality culture, and ensuring product quality amidst raw material variability, contamination risks, and increasing market demand.
Key Challenges in Pharmaceutical QC:
• Regulatory Compliance:
• Evolving Standards: Keeping up with changing global regulations, especially regarding data integrity and validation, is a constant challenge.
• Audit-Readiness: Maintaining operations in a state of readiness for regular audits from regulatory bodies is demanding.
• Data Integrity and Digitalization:
• Data Management: Managing and integrating vast amounts of data from various sources, including manufacturing, labs, and audits, is complex.
• Digitalization Pace: QC systems must adapt to the accelerating speed of digitalization in manufacturing, requiring more integrated reporting and real-time monitoring.
• Quality Culture and Human Factors:
• Cultural Shift: Fostering a robust quality culture requires overcoming resistance to change and a fundamental shift in mindset and employee behavior.
• Employee Engagement: Ensuring active participation, adequate training, and continuous education for all employees is essential to build quality into every step.
• Resource Allocation: Securing sufficient resources for technology, infrastructure, and training can be a significant financial challenge, particularly for smaller companies.
• Operational and Product Quality Challenges:
• Raw Material Variability: Variability in raw materials and their impact on product quality is a persistent difficulty.
• Contamination Risks: Preventing cross-contamination and microbial contamination is a critical challenge, especially for sensitive products like biologics.
• Equipment and Process Management: Managing equipment malfunctions, ensuring proper cleaning, and dealing with legacy systems can impact operational efficiency.
• Supply Chain and Production Demands:
• Complex Supply Chains: Ensuring consistent quality across global supply chains, from raw material sourcing to final product delivery, is a significant task.
• Increased Speed and Efficiency: QC labs face pressure to improve operational efficiency and reduce errors to keep up with growing market demands and faster production cycles.
Pharmaceutical QMS management System
The collaboration of cross-functional teams, often across multiple sites, while ensuring strict compliance with global regulations. Some common pharmaceutical regulations include GMP, FDA 21 CFR Part 210 & 211 (21 CFR Parts 210 and 211 are United States FDA Regulations outlining the cGMP for the drug industry, with Part 210 setting the general framework and definitions, and Part 211 providing detailed requirements for finished pharmaceutical products), 21 CFR Part 11 (Electronic Records and Electronic Signatures requirements used by pharmaceutical or medical device manufacturers when they submit to the FDA), and EU GMP Annex 11 (Manufacture of Sterile Medicinal Products) among others. Any compliance mishaps can lead to a spiral of consequences, from regulatory warnings to product recalls and reputational damage.
This is no easy task for quality professionals from the industry. Maintaining a strong QMS requires balancing complex processes, ensuring high-quality and accurate documentation, and implementing effective risk management strategies, all while keeping operations efficient.
In this article, we’ll explore the most common pharma QMS challenges and provide practical solutions to help your organization stay compliant, improve efficiency, and build a culture of continuous quality improvement.
• Your audit results and management reviews feed into the QMS, enabling continuous improvement
• Establish Quality Policies with clear Quality Objectives in place.
• Your CAPA system helps improve processes continuously, address any audit findings and mitigate risks.
• There is a constant and effective analysis of data: monitoring, measuring, analyzing, and improving processes to ensure sustained quality and hitting those quality KPIs.
Common QMS challenges in pharma
Data integrity and data management
The life sciences industry, including pharma, generates the equivalent of 5 billion pages of text every 24 hours amounting to tens of terabytes of data every single day. As you can imagine, data storage becomes a major concern, as how you store your data impacts costs, security, and resource requirements in significant ways.
Then there’s accessibility, can you quickly pull the documents you need during an audit? If not, you’re in trouble. The lack of data standardization also complicates things.
How can you analyze and use data effectively if it’s inconsistent and scattered across different systems?
Compliance is another big factor. Is your data management in line with regulations like HIPAA? Is your data reliable, accurate, and trustworthy? Nowadays, regulators are increasingly focusing on data integrity to ensure patient safety.
Developing a strong data governance framework, designating a data steward, implementing FAIR principles, and leveraging technologies like AI and ML to analyze large datasets, identify patterns, and predict future trends can help you mitigate some of these challenges.
CAPA and risk management
CAPA failures have been identified as one of the most common audit issues in the pharmaceutical industry in recent years. Inadequate root cause investigations, poor analysis of deviations, product failures, and customer or patient complaints, along with insufficient CAPA systems, have all contributed to these challenges.
Reactive Vs. proactive risk management is common in many pharmaceutical companies as they struggle to continuously monitor outcomes and predict risks.
Build a robust CAPA management system from the start: Use a centralized, digitized system, review it regularly, and ensure it stays current and effective. Establish clear communication guidelines, and make sure each CAPA is reviewed thoroughly with a cross-functional team involved in the process.
Early detection of deviations and nonconformities is crucial for preventing quality issues and driving improvement. What’s the ultimate goal? Prevent these issues in the first place and you can do that through effective risk based thinking and risk management strategies.
Quality culture
Resistance to change, lack of manager buy-in, a blame culture, and quality being seen as just the quality department’s job, sound familiar? These are all signs of a weak quality culture. Every quality assurance manager dreams of a strong, organization-wide quality culture, but sadly, that’s often not the reality.
Building that quality culture is a mindset shift, it requires changing the attitudes of employees used to the status quo, strong support from leadership, and decisive action. This is something that cannot be achieved overnight and is a constant struggle for quality teams as they know their QMS would be so much better if only everybody else was on board.
ISPE’s framework for cultural excellence is built on six key dimensions that drive operational greatness and assess your quality maturity:
1. Building leadership and vision
2. Challenging mindsets and attitudes
3. Active engagement through Gemba walks
4. Leading quality indicators and monitoring key triggers
5. Transparent oversight and review
6. Implementing cultural enablers (e.g., learning organizations, proactive problem-solving, and recognizing organizational change).
Continuous improvement
Quality assurance professionals are often pulled into handling immediate issues; product defects, audits, compliance concerns, or customer complaints. This reactive focus leaves little room for proactive improvement efforts, especially when day-to-day operations and regulatory compliance take priority.
At the end of the day, continuous quality improvement is about teamwork. In organizations with fragmented departments or poor communication, QA managers often struggle to align cross-functional teams around improvement initiatives, making progress slow and difficult.
Adopting one of the tried-and-true methodologies is the Lean Thinking, Six Sigma, and Kaizen.
• Lean Thinking: A continuous transformation that streamlines operations by eliminating waste while maintaining or increasing value.
• Six Sigma: A data-driven method that uses statistical analysis to detect and eliminate inefficiencies and defects, reducing process variation.
• Kaizen: A philosophy of small, incremental improvements that, over time, lead to significant process enhancements.
Supply chain management and compliance
The pharmaceutical industry is one of the most heavily regulated fields worldwide with constantly evolving standards, making compliance across various regions of the supply chain extremely difficult. This means companies must stay on top of continuously updating their processes, documentation, and training programs.
To add to the complexity, the supply chain involves multiple suppliers and manufacturers, which generates gigantic quantities of data to manage and ensure visibility and traceability. And if that wasn’t enough, let’s not forget that the supply chain is vulnerable to disruptions like natural disasters, geopolitical tensions, pandemics, and supply shortages.
Manage a supply chain: by implementing advanced technology solutions. For example, an eQMS can automate repetitive tasks and digitize records, significantly reducing manual errors and improving efficiency. Having suppliers, manufacturers, and distributors aligned on a digital platform also makes it easier to communicate and collaborate.
On the other hand, blockchain technology can further help you enhance traceability by providing a secure, immutable ledger that records every transaction, helping combat counterfeit drugs and ensuring transparency.
Siloed and hybrid quality management
Documentation is the lifeblood of quality management. When it’s managed in a fragmented way, some on paper, others in a mix of digital and paper formats, it leads to disconnected information streams across the organization.
This fragmentation makes it difficult to gain a comprehensive, real-time understanding of processes, making it extremely hard for the quality assurance team to spot potential issues early and react before they escalate. The consequences? Product quality and compliance are compromised, and collaboration becomes inefficient and frustrating.
The best way to unify your documentation and data is by embracing modern technology, like an eQMS. It brings everything together in one secure place, making your processes more efficient and easier to manage.
One of the standout benefits of eQMS is how it seamlessly integrates key QMS functions like document management, risk management, and CAPA, making data extraction for KPIs easy. With all your metrics in one spot, you can quickly spot weak areas, make better decisions, and ensure everyone stays aligned with real-time insights from anywhere.
• Make data work for you, not against you: A strong data governance framework and digital tools will help you ensure integrity, compliance, accessibility, and better decision-making.
• Quality Management should be proactive, not reactive: Strengthening quality management requires a strategic shift towards continuous improvement, proactive risk management, and a deeply ingrained quality culture.
• An eQMS will help ease most of your headaches: A well-integrated eQMS not only reduces manual errors and compliance risks but also provides real-time insights, enabling more informed decision-making and long-term operational success.
• Managing Documentation: Regulatory submissions require extensive and accurate documentation, and manually managing large volumes of data can lead to errors and inefficiencies.
• Talent and Training Gaps: Ensuring that all staff are adequately trained on complex regulations and procedures is crucial but can be challenging.
• Fraud and Corruption: The industry faces risks such as off-label promotion, kickbacks, and bribery, which require stringent controls to prevent.
Strategies to Address Challenges
• Strategic Regulatory Engagement: Early and proactive engagement with regulatory bodies helps streamline the approval process.
• Technological Integration: Utilizing advanced technologies, such as AI and data analytics, can improve data management, transparency, and operational efficiency.
• Robust (QMS): Implementing comprehensive QMS helps monitor and control processes for consistent quality and compliance.
• Employee Empowerment and Training: Investing in thorough and ongoing training programs ensures employees understand and adhere to regulatory requirements.
• Focus on Data Security: Implementing advanced access controls and secure digital communication channels is vital to protect sensitive data.
Edu Journey Mobile App
Online learning now in your fingertips

Sign up to receive our latest updates
Get in touch
Call us directly?
Address:
Email: