
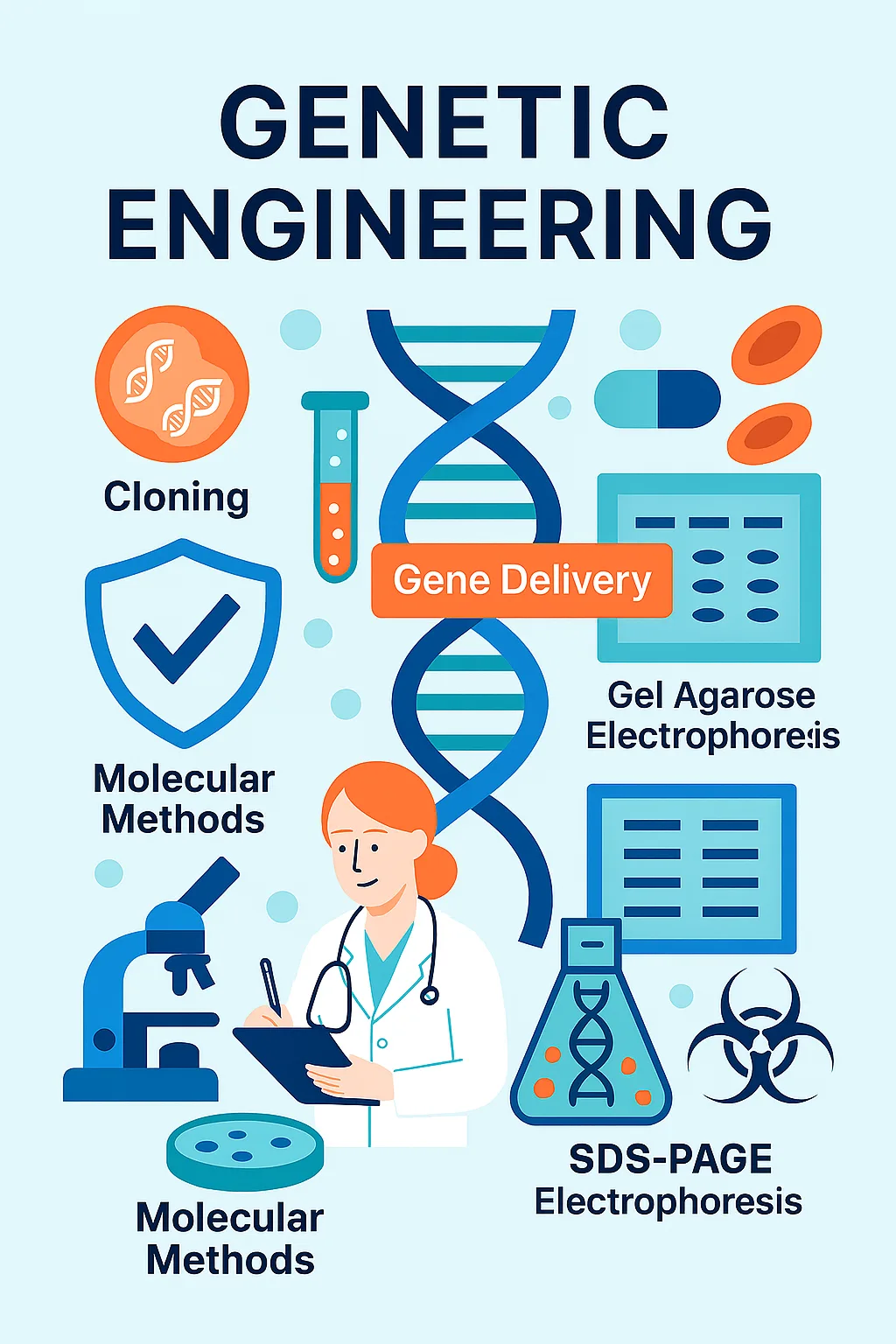
Genetic Engineering
Genetic Engineering
Cloning
Cloning involves molecular cloning (Gene Cloning), which is the process of generating identical copies of a specific DNA sequence (a gene) by inserting it into a host organism, like a bacterium, which then replicates it.
Moreover, it consists of cell line cloning, which is the process of isolating a single cell and allowing it to proliferate in culture to create a population of genetically identical cells, known as a clonal cell line. This application is achieved through various techniques, with the most common being recombinant DNA technology, polymerase chain reaction (PCR), and single-cell dilution cultures. This procedure’s ability to create genetically identical copies has revolutionized biological research and medicine.
The most important application of DNA cloning is used in biopharmaceuticals, gene therapy, and gene analysis.
Cloning procedures involve creating multiple identical copies of a specific DNA fragment, often a gene, using a host organism (mammalian, eukaryotic, and prokaryotic). DNA cloning is the process of making multiple, identical copies of a particular piece of DNA. In a typical DNA cloning procedure, the gene or other DNA fragment of interest is first inserted into a circular piece of DNA called a plasmid. The insertion is done using enzymes that “cut and paste” DNA, and it produces a molecule of recombinant DNA, or DNA assembled out of fragments from multiple sources. This allows for the amplification of the gene and, in some cases, its expression to produce a protein.
Cloning technology faces significant practical challenges, including low-efficiency ligation. That can be overcome by optimizing the insert and using high-concentration T4 DNA Ligase. Restriction enzyme issues that to solve this problem, performing double digestion and optimizing digestion time & conditions are too effective. No colonies or too many empty colonies can be solved by using controls and antibiotic selection & blue-white screening are too effective. PCR-related problems can be solved by using a high-fidelity polymerase and designing effective primers. Toxic inserts can be solved by using tightly regulated expression vectors and cloning at lower temperatures.
Our team offers hands-on training and consulting to troubleshoot cloning experiments, optimize digestion time, and ensure optimal insert orientation. By addressing these challenges, we help researchers and companies achieve reliable, reproducible results efficiently.
Molecular Methods
Molecular biology techniques are common methods used in biology, which generally involve the manipulation and analysis of DNA, RNA, protein, and lipid.
Polymerase chain reaction (PCR)
This is one of the most important techniques used in molecular biology and is basically used to copy DNA. PCR allows a single DNA sequence to be amplified into millions of DNA molecules. PCR can also be used to introduce mutations within the DNA or introduce special restriction enzyme sites. In addition, PCR is used to determine whether a certain DNA fragment exists in a cDNA library. Different types of PCR include reverse transcription PCR (RT-PCR) for amplification of RNA and quantitative PCR (QPCR) to measure the amount of RNA or DNA present.
This procedure has practical applications in forensics, genetic testing, and diagnostics. PCR can also be used to test for a bacterium or DNA virus in a patient’s body: if the pathogen is present, it may be possible to amplify regions of its DNA from a blood or tissue sample.
PCR procedure requires a DNA polymerase enzyme, like DNA replication in an organism that makes new strands of DNA, using existing strands as templates. Two primers are used in each PCR reaction, and they are designed so that they flank the target region (the region that should be copied). The primers bind to the template by complementary base pairing. When the primers are bound to the template, they can be extended by the polymerase, and the region that lies between them will get copied. Steps of PCR consist of Denaturation for separating the double-stranded DNA template strand to the point where the strands start denaturing and the hydrogen bonds are broken between the nucleotide base pairs. Annealing for attachment of the forward and reverse primers to each of the single-stranded DNA template strands. The DNA polymerase is also stable enough to now bind to the primer DNA sequence. Extension is done for the synthesis and elongation of the new target DNA strand accurately and rapidly by DNA polymerase. Final Extension is done to fill in any protruding ends of the newly synthesized strands.
There are many common PCR challenges, such as no amplification that for overcoming this problem, template, primer, Mg²⁺ concentration, and cycling conditions should be checked. Non-specific amplification (multiple bands or smearing) can be overcome by increasing annealing temperature (try a gradient), switching to a hot-start polymerase, reducing primer concentration to 0.2 µM, and using Touchdown PCR. Primer-dimer formation can be overcome by using a hot-start polymerase, reducing primer concentration, and redesigning primers if the problem persists. Low yield (faint band) for overcoming this, increasing the cycle number slightly, increasing the template amount, optimizing annealing temperature (try a gradient), and ensuring extension time is sufficient. False positives in the no-template control (NTC) can be overcome by decontaminating everything and making new aliquots of all reagents.
By understanding these common challenges and their solutions, you can move from frustration to successful and reproducible PCR results. In this line, PCR reaction remains a reliable and versatile method for nucleotide analysis, performing and investigating cloning, and producing high-quality results. For expert consulting or hands-on training in PCR, reach out to our team today.
DNA extraction
DNA extraction method is one of the important steps in the PCR process. DNA samples from different sources are needed to perform a PCR reaction. Various tissues, including blood, body fluids, direct Fine needle aspiration cytology (FNAC) aspirate, formalin-fixed paraffin-embedded tissues, frozen tissue section, etc., can be used for DNA extraction. DNA extraction involves lysing the cells and solubilizing DNA, which is followed by chemical or enzymatic methods to remove macromolecules, lipids, RNA, or proteins. So, the DNA extraction is used to obtain purified DNA by using physical and/or chemical methods from a sample, separating DNA from cell membranes, proteins, and other cellular components. Different procedures for DNA extraction, involving manual and commercially available kits. DNA extraction techniques include organic extraction (phenol–chloroform method), nonorganic method (salting out and proteinase K treatment), and adsorption method (silica–gel membrane).
The general procedure follows three core steps consisting of Lysis for breaking open cells to release DNA, Precipitation for separating DNA from other cellular components (proteins, RNA, lipids) and isolating it, and Purification/Washing for removing salts and other contaminants to get a clean DNA pellet.
The most common challenges consist of low yield of DNA (not enough DNA) that can be overcome by increasing the starting material, optimizing Lysis, and preventing nuclease activity.
Poor Quality/Purity (dirty DNA) is another problem in which the DNA is presented with contamination such as protein, RNA, salt or ethanol, and polysaccharide or humic acid. These contaminants inhibit downstream applications like PCR, restriction digestion, or sequencing. For overcoming this problem, proteinase K, RNase A, column-based kits, and high-salt precipitation are used.
By understanding the biochemical principles behind each step, you can effectively diagnose and solve almost any DNA extraction problem. For expert consulting or hands-on training in DNA extraction, reach out to our team today.
Real-Time PCR
Real-time PCR, also known as quantitative PCR (qPCR), is a molecular biology technique that monitors the amplification of a DNA or RNA sequence during the polymerase chain reaction (PCR) process in real-time, as opposed to at the end of the reaction like in conventional PCR. This allows for the quantification of nucleic acids, making it a valuable tool for various applications. There are important differences between the usual PCR and Real-time PCR.
In real-time PCR, the amplification of DNA or RNA is monitored during each cycle of the PCR process using fluorescent signals. Real-time monitoring enables the quantification of the starting amount of the target nucleic acid, unlike conventional PCR, which only indicates presence or absence. Three general methods are used for the quantitative detection, such as Hydrolysis probes (TaqMan, Beacons, Scorpions), Hybridization probes (Light Cycler), and DNA-binding agents (SYBR Green).
Important agents in Real-time PCR is related to Fluorescent Detection: Real-time PCR utilizes fluorescent dyes or probes that bind to the amplified DNA, generating a fluorescent signal that is proportional to the amount of DNA present. Cycle Threshold (Ct): The point at which the fluorescence signal crosses a threshold (Ct value) is used to determine the initial amount of target DNA. A lower Ct value indicates a higher starting concentration of the target. Data Analysis: The fluorescence data collected during each PCR cycle are used to generate amplification curves, allowing for quantification of the target DNA.
The applications of Real-time PCR consist of gene expression analysis, pathogen detection, genetic testing, food safety, and research studies.
Real-time PCR procedure begins with the meticulous preparation of a reaction mix containing the template DNA (or reverse-transcribed cDNA for RT-qPCR), sequence-specific forward and reverse primers, a fluorescent reporter (typically either a DNA-binding dye like SYBR Green or a sequence-specific fluorescent probe, such as a TaqMan probe), nucleotides (dNTPs), a buffer with magnesium ions, and a thermostable DNA polymerase. This mixture is aliquoted into individual wells of a specialized optical plate or tube, which is then sealed and placed into the thermal cycler of the real-time PCR instrument. The instrument then executes a programmed series of temperature cycles—initial denaturation, followed by repeated cycles of denaturation, annealing, and extension—while simultaneously exciting the fluorescent dyes with a light source and measuring the intensity of the emitted fluorescence at the end of each cycle. As the target sequence is exponentially amplified, the accumulating product binds the fluorescent dye, causing a measurable increase in fluorescence signal that is directly proportional to the amount of amplicon generated. This data is collected in real-time by the instrument’s software, which subsequently plots fluorescence against cycle number to generate an amplification curve for each sample, allowing for the precise quantification of the initial target amount through the determination of the cycle threshold (Ct) value, where the fluorescence exceeds a background level.
Real-Time PCR is a powerful technique, but its sensitivity makes it prone to issues. Problems generally fall into a few categories: amplification issues, signal issues, data quality issues, and contamination. Amplification issues related to No Ct, Late Ct, or failed reactions. This is the most common problem in Real-Time PCR that the reaction either doesn’t amplify at all or amplifies very inefficiently. To solve this problem, the quality and degradation of the template and presentation of inhibitors such as phenol, heparin, ethanol, and salts should be checked. Moreover, primer or probe issues, which consist of wrong design, degradation, or incorrect concentration, can be solved by checking BLAST sequences to ensure the specificity and comparing the concentrations of the primer can be useful. Signal & Fluorescence issues are other problems related to the detection of the fluorescent signal. Low fluorescence signal is an important problem that creates a weak signal in even positive controls and shows high background noise. To solve this problem, keeping probes in the dark (use amber tubes) and avoiding repeated freeze-thaw cycles are highly recommended. Data quality & reproducibility issues with the reaction runs, in which the data is inconsistent or unreliable, are related to pipetting error and template inhomogeneity. Using quality pipettes, calibrating them, and mixing the template thoroughly by vortexing and spinning down all samples and master mix before use are effective ways.
By following this structured approach, investigating general troubleshooting, you can systematically diagnose and resolve most qPCR challenges. In this line, our team offers expert consulting or hands-on training in Real-Time RCR.
RNA Extraction
RNA isolation is an important step in analysing gene expression, which is a common application of RT-qPCR. Gene expression requires RNA, which is reverse transcribed into cDNA, and the cDNA in turn becomes the template for the qPCR amplification in an experiment. Hence, the first step in gene expression analysis is RNA isolation.
There are four common RNA extraction methods, which consist of Organic (Phenol-Chloroform) Extraction (e.g., TRIzol®, TRI Reagent®). Spin Column (Silica Membrane) Based Purification, Magnetic Bead-Based Purification, and Automated Extraction Systems.
In essence, RNA extraction procedure is a careful balancing act consisting of breaking open cells and tissue, inactivating RNases to prevent degradation, separating RNA from DNA, proteins, and other cellular components, washing away all contaminants, eluting the pure, intact RNA in a stable solution, and verifying its quality and quantity before use.
On the other hand, the disadvantages and limitations of RNA extraction are related to rigorous RNase inhibition, RNA instability, and co-purification of inhibitors.
RNA Extraction method is vital for Gene expression analysis, a diagnosis of RNA viruses, Research and discovery of novel RNA species, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), functional genomics, which provides a dynamic view of cellular activity, and Next-Generation Sequencing (NGS) for RNA-Seq.
The most common challenges are related to RNase contamination, RNA degradation, genomic DNA (gDNA) contamination, and low yield.
As RNases are ubiquitous and extremely robust, they don’t denature by autoclaving and can renature after heating. Using RNase-free reagents, consumables, and equipment, wearing gloves and changing them frequently, and using Diethyl pyrocarbonate (DEPC) to inactivate RNases, are highly recommended. The result of RNase activity leads to smeared and degraded RNA bands on an electrophoresis gel instead of sharp ribosomal RNA bands. So, to solve this issue, checking the quality of RNA by running it on a gel and using the correct lysis buffer-to-sample ratio to ensure immediate inactivation of RNases is necessary.
As DNA contamination can co-precipitate with RNA, especially if using silica-column methods without a DNase step. This causes false positives in qPCR. In this line, the gold standard is performing an on-column DNase I digestion step.
Furthermore, low yield during RNA extraction can be modified by starting with more sample and ensuring the tissue is thoroughly homogenized to liberate all RNA.
Mastering this technique is a cornerstone of modern molecular biology and genetics research. So, our team has hands-on experience in addressing each of these pain points. We provide consulting services for improving RNA stability and purity, training in advanced preparation techniques, and strategic guidance for scale-up and regulatory compliance. By tackling laboratory challenges, we can ensure RNA extraction products reach their full potential for other molecular techniques.
Gel Agarose Electrophoresis
Gel agarose electrophoresis is a fundamental laboratory technique used to separate DNA or RNA fragments based on size. It provides a simple yet powerful method for analyzing nucleic acids, verifying PCR products, and checking sample integrity. Its importance lies in providing reliable information on nucleic acid quality and fragment size; without proper execution, results can be smeared, faint, or misleading, impacting downstream experiments.
The advantages of agarose gel electrophoresis include its simplicity, cost-effectiveness, and rapid visualization of nucleic acids. It allows researchers to quickly assess the success of amplification or restriction digestion and is widely used in molecular biology, genetics, and diagnostics.
Despite its simplicity, laboratories often face practical challenges. Uneven gel polymerization or improper agarose concentration can cause distorted bands, while sample overloading may lead to smearing. Poor quality or degraded samples can reduce signal intensity. These issues can be addressed by optimizing gel concentration, using high-quality buffers, controlling sample amounts, and applying standardized running conditions. Additionally, safety concerns related to ethidium bromide or other dyes require proper handling and disposal protocols.
Our team offers hands-on training and consulting to troubleshoot agarose electrophoresis experiments, optimize gel and buffer compositions, and implement safer staining techniques. By addressing these challenges, we help researchers and companies achieve reliable, reproducible results efficiently.
Overall, agarose gel electrophoresis remains an essential tool for nucleic acid analysis, and expert guidance can maximize both accuracy and efficiency in laboratory workflows. For practical training or consulting in agarose gel electrophoresis, contact our team today.
SDS-PAGE Electrophoresis
SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) is a widely used technique to separate proteins based on their molecular weight. By denaturing proteins and imparting uniform negative charge, SDS-PAGE enables precise protein profiling, purity assessment, and verification of protein expression. Its importance is critical: incorrect gel preparation or running conditions can lead to distorted bands, incomplete separation, or inaccurate molecular weight estimation.
The advantages of SDS-PAGE include high resolution, reproducibility, and adaptability to downstream applications such as Western blotting or protein quantification. It is a cornerstone technique in proteomics, drug discovery, and biotechnology research.
Laboratories often encounter several challenges. Incomplete protein denaturation or sample aggregation can affect separation quality. Gel polymerization errors or improper acrylamide percentage can distort migration patterns. Additionally, overheating during electrophoresis may cause band smearing, while low-quality reagents reduce resolution. These problems can be solved by using optimized sample preparation protocols, precise gel casting techniques, and controlled electrophoresis conditions. Our consulting services guide labs in selecting the correct gel percentage for protein sizes, preparing high-quality reagents, and troubleshooting experimental anomalies.
SDS-PAGE remains a reliable and versatile method for protein analysis, and proper guidance ensures reproducible, high-quality results. For expert consulting or hands-on training in SDS-PAGE electrophoresis, reach out to our team today.
Western Blotting
Western blotting is a powerful analytical technique used to detect specific proteins in complex samples. By combining SDS-PAGE separation with antibody-based detection, it provides qualitative and quantitative insights into protein expression, post-translational modifications, and biomarker validation. Its importance is paramount: improper transfer, antibody selection, or blocking can produce weak or non-specific signals, compromising experimental conclusions.
Western blotting offers significant advantages over other protein detection methods, including high specificity, versatility in sample type, and compatibility with multiple detection systems (chemiluminescence, fluorescence, or colorimetric). Applications range from basic research in cell biology to clinical diagnostics, drug development, and biomarker discovery.
Despite its widespread use, real-world challenges are common. Inefficient protein transfer can occur due to incorrect membrane choice or running conditions. Non-specific antibody binding or high background signal often results from insufficient blocking or suboptimal antibody concentrations. Reproducibility issues may arise from inconsistent sample loading or variable gel quality. These can be addressed through careful selection of membranes and antibodies, standardized blocking protocols, and optimized transfer and detection conditions. Our team provides specialized consulting and hands-on workshops to troubleshoot these issues, improve signal quality, and ensure reproducible results.
Western blotting continues to be an indispensable tool for protein analysis, and professional guidance can significantly enhance both accuracy and reliability. For training or consulting in Western blotting, our expert team is ready to support your laboratory’s needs.
Edu Journey Mobile App
Online learning now in your fingertips

Sign up to receive our latest updates
Get in touch
Call us directly?
Address:
Email: